فهرست مطالب
سندرم پرادرویلی (Prader-Willi syndrome) یک اختلال ژنتیکی نادر است که منجر به مشکلات جسمی، ذهنی و رفتاری میشود. یکی از ویژگیهای اصلی سندرم پرادرویلی احساس گرسنگی مداوم است که بهطور معمول در حدود 2 سالگی شروع میشود. ما در ادامه به این موضوعات میخواهیم بپردازیم که سندرم پرادرویلی چیست؟ علائم سندرم پرادرویلی به چه صورت است؟

سندرم پرادرویلی چیست؟
سندرم پرادرویلی یک بیماری ژنتیکی پیچیده است که بسیاری از قسمتهای بدن را تحت تاثیر قرار میدهد. در دوران نوزادی، این وضعیت با تون (tone) عضلانی ضعیف، مشکلات تغذیهای، رشد ضعیف و تاخیر در رشد مشخص میشود. افراد مبتلا در دوران کودکی دچار گرسنگی شدید میشوند که منجر به پرخوری مزمن و چاقی میشود. برخی از افراد مبتلا به سندرم ”Prader-Willi“ (به ویژه آنهایی که چاق هستند)، به دیابت نوع 2 نیز مبتلا میشوند.
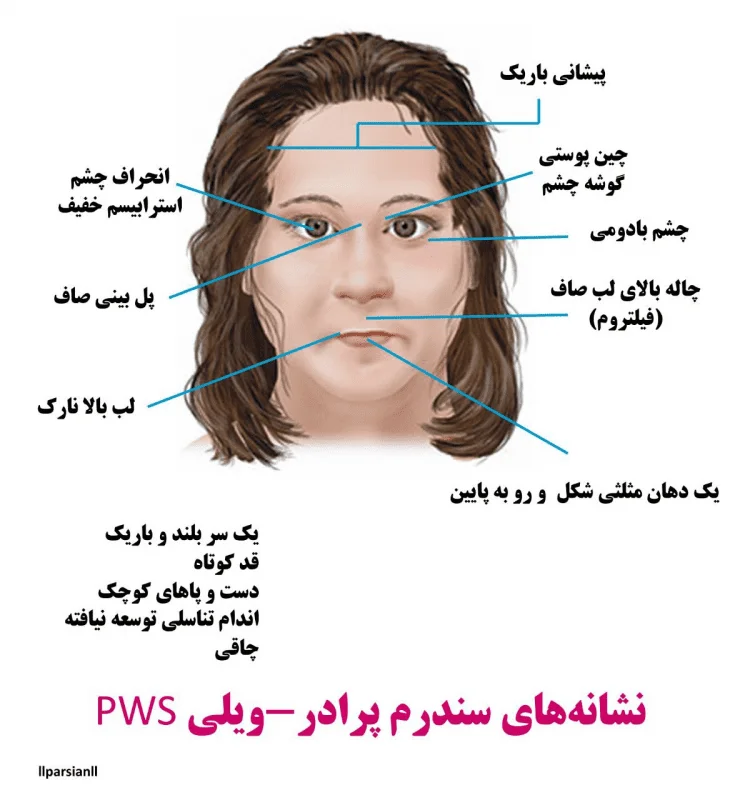
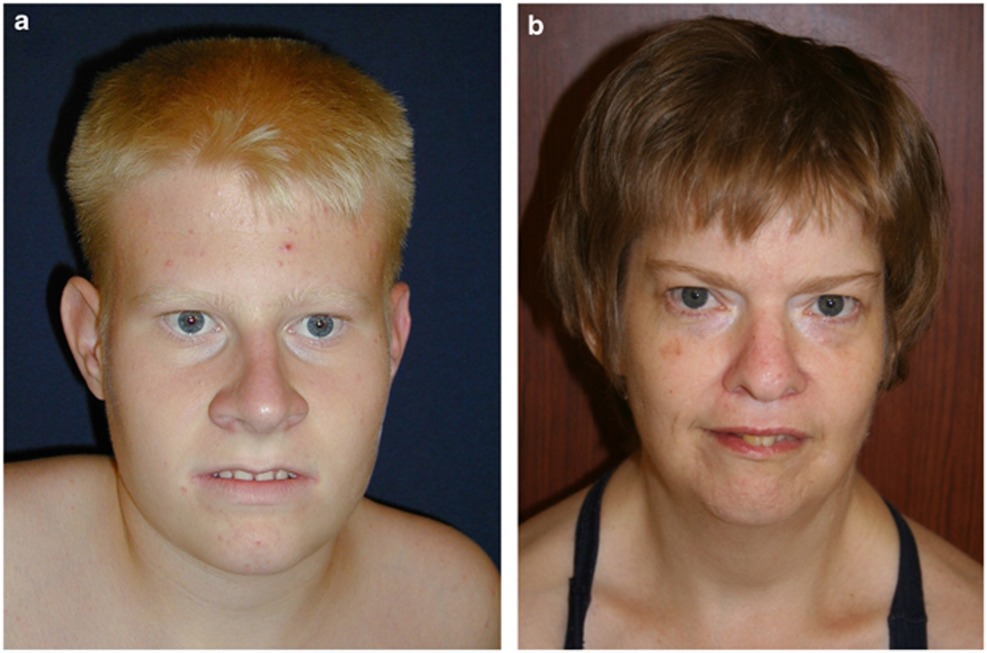
افراد مبتلا به سندرم پرادرویلی بهطور معمول دارای اختلالات ذهنی خفیف تا متوسط و ناتوانیهای یادگیری و مشکلات رفتاری مانند لجبازی هستند. اختلالات خواب نیز ممکن است رخ دهد. ویژگیهای اضافی این بیماری شامل ویژگیهای متمایز صورت مانند چشمهای بادامی شکل، پیشانی باریک، قد کوتاه، دهان مثلثی و دستها و پاهای کوچک است. برخی از افراد مبتلا به سندرم پرادرویلی دارای پوست غیرعادی روشن و موهای روشن هستند. مردان و زنان مبتلا به سندرم پرادرویلی اندام تناسلی توسعه نیافته دارند. بلوغ تاخیری یا ناقص است و اکثر افراد مبتلا به سندرم Prader-Willi قادر به بچهدارشدن نیستند.
اپیدمیولوژی (Epidemiology)سندرم پرادرویلی
سندرم پرادرویلی شیوع 1 در هر 20000 تا 1 در هر 30000 تولد دارد. مبنای اصلی تشخیص، آزمایش متیلاسیون “DNA” برای شناسایی هرگونه نقص در “imprinting”والدین بر روی کروموزوم 15 است و این آزمایش میتواند بیش از 99 درصد افراد مبتلا را تشخیص دهد. با این وجود، در اکثر موارد در سن 3.9 سالگی حتی در مراکز مرجع تشخیص داده میشوند. در سطح جهان، بهطور تقریبی 400000 نفر مبتلا به سندرم prader-williهستند.
علائم سندرم پرادرویلی
علائم معمول سندرم پرادرویلی شامل موارد زیر هستند:
- اشتهای بیش از حد و پرخوری که بهراحتی میتواند منجر به افزایش وزن خطرناک شود؛
- رشد محدود (کودکان بسیار کوتاهتر از حد متوسط هستند)؛
- شلشدن ناشی از ضعف عضلات؛
- مشکلات یادگیری؛
- عدمرشد جنسی؛
- چالشهای رفتاری مانند طغیان عاطفی و پرخاشگری فیزیکی.
پاتوفیزیولوژی (Pathophysiology) سندرم پرادرویلی
در بدو تولد، شاخصهای رشد مانند وزن، طول و شاخص توده بدنی (body mass index) در بیماران مبتلا به سندرم پرادرویلی 15 تا 20 درصد کمتر از خواهر و برادر سالم آنها است که نشان میدهد رشد در دوران بارداری نامنظم است.
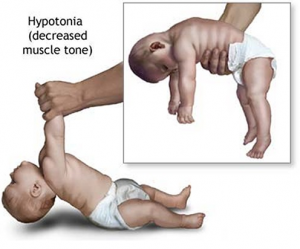
هیپوتونی (Hypotonia) قبل از تولد باعث کاهش حرکت جنین میشود، وضعیت غیرطبیعی در زمان زایمان منجر به افزایش سزارین و زایمان کمکی میشود.
تظاهرات بالینی سندرم پرادرویلی
هیپوتونی: هیپوتونی نوزادی بسیار مشخص است و بهطور تقریبی در تمام افراد مبتلا به سندرم پرادرویلی دیده میشود. آنها دارای رفلکس مکیدن ضعیفی هستند که مشکلات تغذیهای ایجاد میکند، تودهی عضلانی و قدرت آنها کاهش مییابد. در اوایل زندگی رشد نمیکنند. نیاز به داشتن تجهیزات ویژهای برای تغذیه یا نیاز به گاواژ (gavage) برای بهبود تغذیه برای مدت طولانی دارند.
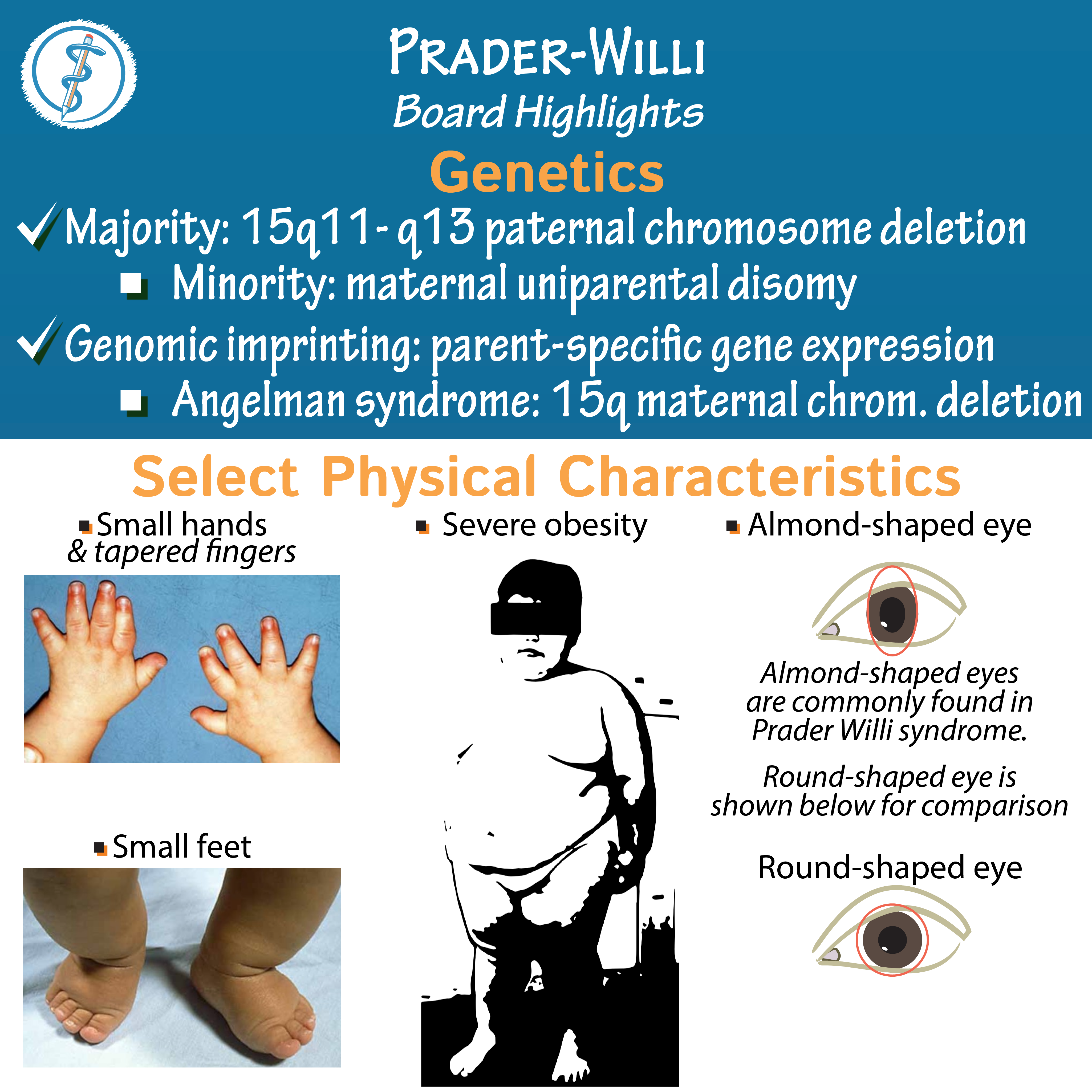
ویژگیهای بدشکلی: برخی از ویژگیها صورت شامل “almond-shaped palpebral fissures”، قطر پیشانی باریک، استرابیسم (strabismus)، پل بینی باریک، گوشههای رو به پایین دهان و هیپوپلازی (hypoplasia) مینای دندان است. آنها همچنین میتوانند با دستها و پاهای کوچک ظاهر شوند.
تاخیر در رشد و مشکلات رفتاری: تاخیر رشد حرکتی در اکثر افراد مبتلا به سندرم پرادرویلی وجود دارد. اختلال زبان نیز بهطور معمول بهعنوان ناتوانیهای فکری و یادگیری دیده میشود؛ اما زمانیکه فرد به سن مدرسه میرسد، مشهودتر است. بهطور تقریبی تمام موارد سندرم پرادرویلی با درجاتی از مشکلات رفتاری مانند اختلال وسواس فکری-اجباری، اضطرابی، طغیانهای خلقی و آسیبهای ناشی از خود همراه است.
کمبود هورمون رشد: کوتاهی قد میتواند از اوایل کودکی وجود داشته باشد و بهطور تقریبی در همهی موارد در دههی دوم زندگی ظاهر میشود. کمبود هورمون رشد شایعترین نقص غدد درونریز است. اگرچه بیماریزایی بهطور کامل مشخص نیست؛ “IGF-1” بهطور تقریبی در هر بیمار مبتلا به سندرم پرادرویلی کمبود دارد یا غیرطبیعی است.
علل سندرم پرادرویلی
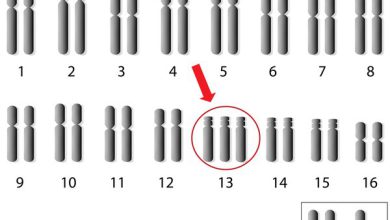
ژنها حاوی دستورالعملهایی برای ساخت یک انسان هستند که از DNA تشکیلشدهاند و در رشتههایی بهنام کروموزوم بستهبندی میشوند. یک فرد 2 نسخه از تمام ژنهای خود دارد که به این معنی است که کروموزومها بهصورت جفت هستند. انسان 46 کروموزوم دارد. یکی از کروموزومهایی که متعلق به جفت شمارهی 15 است در سندرم پرادرویلی متفاوت است.
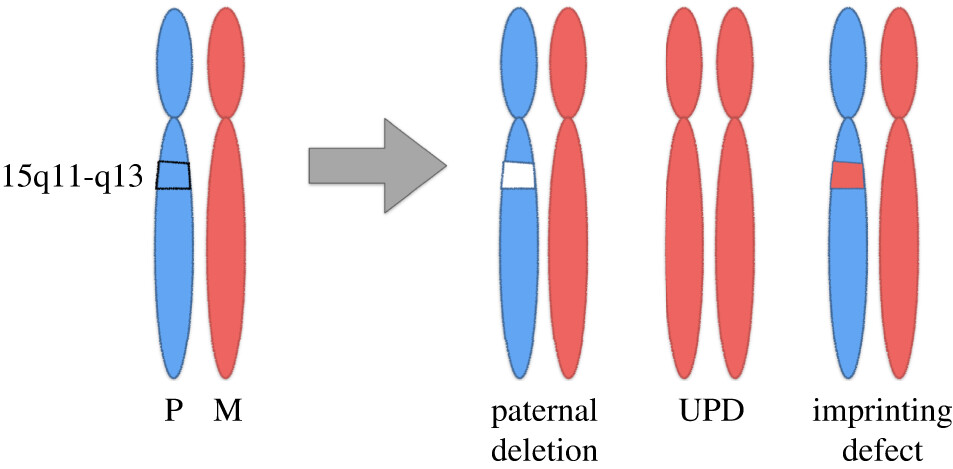
حدود 70 درصد از موارد سندرم پرادرویلی نتیجهی از دسترفتن اطلاعات ژنتیکی از کپی کروموزوم 15 است که از پدر به ارث میرسد. این بهعنوان «حذف پدری» (paternal deletion) نامیده میشود. تصور میکنند که حذف پدری بهطور کامل تصادفی اتفاق میافتد؛ بنابراین وجود بیش از 1 کودک مبتلا به سندرم پرادرویلی که ناشی از حذف کروموزوم پدر است، غیرمعمول است. با این حال، اگر سندرم پرادرویلی به دلیل نوع دیگری از تغییر در کروموزوم 15 رخ دهد، احتمال بسیار کمی وجود دارد که فرزند دیگری با این سندرم متولد شود.
سندرم پرادرویلی در 70 درصد موارد بهدلیل از دسترفتن بیان ژن در ژنهای پدری بهارثرسیده در کروموزوم “15q11.2-q13” ناشی از اشتباهات imprintingبهعلت حذف پدری رخ میدهد و در 25 درصد موارد دیگر نقص بهدلیل دیزومی تک والدی مادری (maternal uniparental disomy) اتفاق میافتد.
سندرم پرادرویلی بهدلیل کمبود مواد ژنتیکی در گروهی از ژنهای کروموزوم شمارهی 15 ایجاد میشود. این امر منجر به مشکلاتی میشود که بخشی از مغز به نام هیپوتالاموس را تحت تاثیر قرار میدهد. هیپوتالاموس هورمون تولید میکند و از این طریق رشد و اشتها را تنظیم میکند.
این ممکن است برخی از ویژگیهای معمول سندرم پرادرویلی مانند تاخیر در رشد و گرسنگی مداوم را توضیح دهد. علت ژنتیکی بهطور کامل تصادفی است و پسران و دختران را از هر قومیتی ممکن است تحت تاثیر قرار دهد. بهندرت ممکن است که والدین، بیش از یک فرزند مبتلا به سندرم Prader-Willi داشته باشند.
تاثیر سندرم پرادرویلی بر روی مغز
مشکل کروموزوم 15 باعث اختلال در رشد و عملکرد بخشی از مغز به نام هیپوتالاموس میشود. هیپوتالاموس در بسیاری از عملکردهای بدن مانند تولید هورمون و کمک به اشتها نقش دارد. اگر هیپوتالاموس بهدرستی عمل نکند ممکن است برخی از ویژگیهای معمول سندرم پرادرویلی مانند تاخیر در رشد و گرسنگی مداوم را توضیح دهد.
مطالعاتی که با استفاده از فناوری پیشرفتهی تصویربرداری مغز صورت گرفتهاست، نشان میدهد که افراد مبتلا به سندرم Prader-Willi پس از غذاخوردن، فعالیت الکتریکی بسیار بالایی در بخشی از مغز به نام قشر پیشانی (frontal cortex) دارند. این قسمت از مغز با لذت جسمانی و احساس رضایت همراه است. ممکن است افراد مبتلا به سندرم پرادرویلی عمل غذاخوردن را بسیار مفید بدانند. فرضیهی دیگر این است که در سندرم پرادرویلی، هیپوتالاموس نمیتواند سطح غذا را در بدن بهدرستی قضاوت کند؛ این بدان معناست که فرد مبتلا به سندرم پرادرویلی بدون توجه به مقدار غذایی که میخورد، همیشه احساس گرسنگی میکند.
تشخیص سندرم پرادرویلی
سندرم پرادرویلی بهطور معمول با آزمایشات ژنتیکی قابل تشخیص است. آزمایش ژنتیک (گاهی به آن آزمایش ژنومی میگویند) تغییراتی را در ژنها پیدا میکند که میتواند باعث مشکلات سلامتی شود. این بهطور کلی برای تشخیص بیماریهای نادر، ارثی و برخی سرطانها استفاده میشود.
اگر کودکی علائم سندرم پرادرویلی را داشته باشد، ممکن است آزمایش ژنتیک توصیه شود. نوزادانی که هنگام تولد بسیار کمجان هستند نیز ممکن است مورد آزمایش قرار گیرند.
چرا آزمایش ژنتیک به فردی پیشنهاد شود؟
- پزشک فکر میکند که ممکن است فرد به یک وضعیت سلامتی ناشی از تغییر در یک یا چند ژن (مثل سندرم پرادرویلی) مبتلا باشد.
- یکی از اعضای خانواده یک وضعیت سلامتی دارد که ناشی از تغییرات ژنی است.
- برخی از بستگان نزدیک نوع خاصی از سرطان داشتهاند که میتواند ارثی باشد.
- فرد یا شریک زندگی آن شخص مبتلا به یک بیماری است که ممکن است به فرزندان منتقل شود.
مدیریت سندرم پرادرویلی
هیچ درمانی برای سندرم پرادرویلی وجود ندارد، بنابراین هدف از درمان، مدیریت علائم و مشکلات مرتبط با آن است. این شامل مدیریت اشتهای بیش از حد فرد و چالشهای رفتاری است. یکی از مهمترین بخشهای مراقبت از فرد مبتلا به سندرم پرادرویلی، تلاش برای حفظ وزن طبیعی است.
آنها باید از همان ابتدا یک رژیم غذایی متعادل داشته باشند و از غذاهای شیرین و پرکالری اجتناب کنند. اگر فرد مبتلا به سندرم Prader-Willi اجازه داشته باشد هر چقدر که میخواهد غذا بخورد، بهسرعت و بهطور خطرناکی اضافهوزن پیدا میکند.
فرد مبتلا به سندرم پرادرویلی میتواند بسیار بیشتر از دیگران غذا بخورد و همچنان احساس گرسنگی کند. محدودکردن مصرف غذا میتواند چالشبرانگیز باشد. افراد مبتلا به سندرم پرادرویلی ممکن است زمانی که غذای اضافه بخواهند، ناامید شوند و به همین دلیل غذا را مخفی کنند یا آن را بدزدند.
هورمون رشد در بیماران مبتلا به سندرم پرادرویلی در اوایل زمان تشخیص از 3 تا 6 ماهگی توصیه میشود. بیمارانی که در دوران کودکی تحت درمان قرار میگیرند، میتوانند به قد نهایی پیشبینیشدهی خود در بزرگسالی برسند.
هومون گنادوتروپیک جفتی انسانی (Human chorionic gonadotropic hormone) یا (hCG) برای کمک به بیماران در پایین آوردن موقعیت بیضه استفاده میشود، اگرچه درصد قابلتوجهی از آنها به ارکیوپکسی (orchiopexy) نیاز دارند.
تستوسترون (testosterone) برای بیماران 15 تا 16 ساله با بلوغ تاخیری یا نقص برای پیشرفت بلوغ تایید شدهاست.
زنان ممکن است به مدت دو سال یا تا زمان قاعدگی تحت درمان با تکههای تراپوستی (transdermal) با دوز پایین برای هیپوگنادیسم (hypogonadism) قرار گیرند.
مشکلات طولانیمدت ناشی از سندرم پرادرویلی
سندرم پرادرویلی تهدیدکنندهی زندگی نیست؛ اما افزایش وزن میتواند باعث شود که بزرگسالان جوان مبتلا به سندرم پرادرویلی به شرایط جدی مرتبط با چاقی مانند موارد زیر دچار شوند:
- دیابت تیپ 2؛
- نارسایی قلبی؛
- مشکلات تنفسی.
اگر رژیم غذایی آنها بهخوبی کنترل شود و دچار اضافهوزن نشوند، بزرگسالان میتوانند کیفیت زندگی خوب و امید به زندگی طبیعی داشته باشند. بسیاری از بزرگسالان مبتلا به سندرم Prader-Willi در فعالیتهایی مانند کار داوطلبانه یا نیمهوقت شرکت میکنند؛ اما بعید است که زندگی بهطور کامل مستقلی داشته باشند.
نویسنده: ریحانه ارفعی
ویراستار: حدیث پرهیزگاری
منابع:
1. https://medlineplus.gov/genetics/condition/prader-willi-syndrome/
2. https://www.nhs.uk/conditions/prader-willi-syndrome/
3. https://www.mayoclinic.org/diseases-conditions/prader-willi-syndrome/symptoms-causes/syc-20355997
4. https://www.ncbi.nlm.nih.gov/books/NBK553161/