فهرست مطالب
سندرم انجلمن (Angelman syndrome) یک اختلال ژنتیکی پیچیده است که در درجهی اول بر سیستم عصبی تاثیر میگذارد. از ویژگیهای بارز سندرم انجلمن میتوان به تاخیر در رشد، ناتوانی ذهنی، اختلال شدید در گفتار و مشکلات حرکتی-تعادلی اشاره کرد. در ادامه میخواهیم به این موضوعات بپردازیم که سندرم انجلمن چیست؟ علائم و درمان Angelman syndrome به چه صورت است؟
سندرم انجلمن چیست؟
Angelman syndrome یک اختلال ژنتیکی است که در درجهی اول بر سیستم عصبی تاثیر میگذارد. سندرم انجلمن توسط دکتر هری انجلمن (Dr. Harry Angelman) در سال 1965 گزارش شد؛ به همین دلیل این اختلال را به افتخار او به این نام خواندند.
سندرم انجلمن در یک نفر از 12000 تا 20000 نفر مشاهده میشود و با تاخیر شدید رشد، اختلال در گفتار، آتاکسی (ataxia) و لرزش اندامها مشخص میشود و دارای رفتار منحصربهفردی هستند که شامل خندیدن مکرر است. علاوهبر این، میکروسفالی (microcephaly) و تشنج در سندرم انجلمن شایع هستند.
نوزادان همراه با سندرم انجلمن در بدو تولد طبیعی به نظر میرسند؛ اما اغلب در ماههای اول زندگی مشکلات تغذیهای دارند. آنها تاخیر در رشد را بین سنین 6 تا 12 ماهگی نشان میدهند. تشنج اغلب بین 2 تا 3 سالگی شروع میشود.

علائم سندرم انجلمن
- تاخیر در رشد؛
- ناتوانی ذهنی؛
- اختلال گفتاری شدید؛
- مشکلات حرکتی-تعادلی (آتاکسی) یا (ataxia)؛
- تشنجهای مکرر (صرع)؛
- رفتار بسیار شاد با خندههای مکرر؛
- اندازهی کوچک سر یا میکروسفالی (microcephaly).
در Angelman syndrome بهطور معمول مشکلات گوارشی، ارتوپدی و چشم وجود دارد؛ همچنین بیشفعالی و توجه کوتاهمدت شایع هستند.
علت بروز سندرم انجلمن
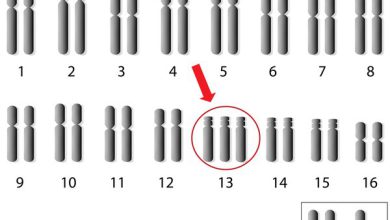
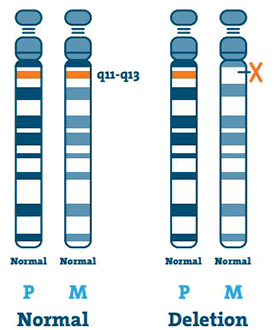
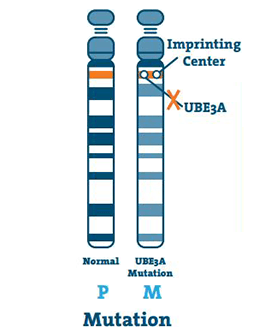
سندرم انجلمن توسط مکانیسمهای مولکولی متفاوتی ایجاد میشود که در نهایت منجر به از دستدادن عملکرد ژن “UBE3A” به ارث بردهشده در ناحیهی کروموزومی “15q11-q13” میشود. این ناحیهی کروموزومی توسط نقشگذاری ژنومی (genomic imprinting) تنظیم میشود و آلل UBE3A در نورونها با مکانیزم خاصی خاموش میشود و این امر از طریق “RNA” آنتیسنس غیرکدکننده “SNHG14” صورت میپذیرد که در گذشته بهعنوان “UBE3A-ATS” نامیده میشد.
ژن UBE3A یک یوبیکوئیتین پروتئین لیگاز “E3A” (E3A ubiquitin ligase protein) با وزن 100 کیلو دالتون را کد میکند که بهعنوان پروتئین مرتبط با “E6” یا بهسادگی UBE3A شناخته میشود.
UBE3A متعلق به کلاس “HECT” یوبیکوئیتین لیگاز “E3” است که بهواسطهی شناسایی پروتئینهای هدف برای تجزیه در پروتئازوم شناختهشدهاست. خاموششدن یا بیان آلل UBE3A مادری جهشیافته در نورونهای بیماران سندرم انجلمن سبب میشود که پروتئین عملکردی UBE3A وجود نداشته باشد؛ در نتیجه، این امر سبب تجمع اهداف UBE3A میشود.
از دستدادن بیان UBE3A در سندرم انجلمن بهطور تقریبی در تمام نورونهای سیستم عصبی مرکزی رخ میدهد. با این وجود، تشخیص اینکه کدام یک از مناطق خاص مغز (هیپوکامپ (hippocampus)، کورتکس (cortex) و مخچه (cerebellum)) سهم خاصی را در سندرم انجلمن ایفا میکند، دشوار است.
تشخیص سندرم انجلمن
معیارهایی برای تشخیص بالینی سندرم انجلمن در ارتباط با کمیتهی مشاورهی علمی بنیاد Angelman syndrome ایالات متحده ایجاد شدهاست. نوزادان بهطور معمول یک فنوتیپ طبیعی دارند. تاخیر در رشد برای اولین بار در 6 ماهگی مشاهده میشود. با این حال، ویژگیهای بالینی منحصربهفرد سندرم انجلمن تا بعد از 1 سالگی آشکار نمیشود و ممکن است چندین سال طول بکشد تا تشخیص بالینی صحیح آشکار شود. تشخیص در ابتدا براساس فنوتیپ رفتاری، اختلال حرکتی، عدم تکلم و رفتار شاد کودک صورت میگیرد. اگرچه برخی از افراد مبتلا به سندرم انجلمن ممکن است بدشکلی خفیف جمجمه-صورت داشته باشند.
آزمایش ژنتیک مولکولی
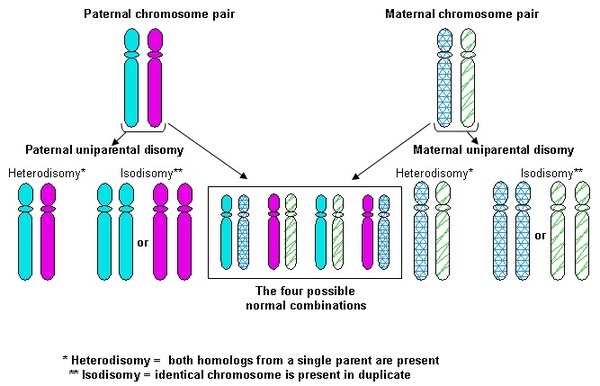
افرادی که مبتلا به Angelman syndrome نیستند، دارای یک آلل “SNRPN” متیله و غیرمتیله در هر دو تجزیهوتحلیل ساترن بلات (Southern blot) و روش زنجیرهای پلیمراز (PCR) خاص متیلاسیون هستند. در افراد مبتلا به سندرم انجلمن یا دیزومی تک والدی (uniparental disomy) “UPD”حذف 5 تا 7 مگابایتی “15q11.2-q13”، در آنها رخ دادهاست؛ همچنین در “ID” (imprinting defect) فقط یک سهم غیرمتیله دارند (اثر متیلاسیون غیرطبیعی والدین).
اکثر آزمایشهای آنالیز متیلاسیون “DNA” تجاری موجود نمیتوانند بین سندرم انجلمن ناشی از حذف، UPD و ID تمایز قائل شوند. روشهای جدیدتر شامل توالییابی پیروفسفاتی (pyrosequencing)، توسعهی پروب وابسته به اتصال چندگانهی اختصاصی متیلاسیون (methylation-specific multiplex ligation-dependent probe amplification)، تجزیهوتحلیل مبتنی بر توالی و سایر روشهای آنالیز شمار نسخه (copy-number analysis) ممکن است بهزودی کمیت کافی را برای تمایز حذفها، UPD و ID فراهم کنند.
تجزیهوتحلیل متیلاسیون DNA تقریبا 80 درصد از افراد مبتلا به سندرم انجلمن را شناسایی میکند و بهطور معمول اولین آزمایش سفارشی است. اگر تجزیهوتحلیل متیلاسیون DNA غیرطبیعی باشد، مرحلهی بعدی آنالیز “FISH” یا “CGH” است. اگر تجزیهوتحلیلهای FISH یا CGH نرمال باشد، تجزیهوتحیلیل پلیمورفیسمهای DNA روی کروموزوم 15 میتواند بین UPD و ID تمایز قائل شود. اگر حذف UPD وجود نداشتهباشد، مطالعات بیشتر میتواند تعیین کند که آیا حذف “IC” وجود دارد یا خیر. اگر متیلاسیون DNA نرمال باشد، آنالیز توالی UBE3A تست تشخیصی مناسب بعدی است.
مدیریت و درمان سندرم انجلمن
مشکلات تغذیهای نوزادان ممکن است به نوک سینههای ویژه و سایر راهکارها برای مدیریت مکیدن ضعیف یا ناهماهنگ نیاز داشته باشد. رفلاکس معده میتواند با افزایش وزن خفیف و استفراغ همراه باشد. درمان طبی مرسوم (وضعیت عمودی و داروهای حرکتی) بهطور معمول موثر است. گاهی فوندوپلیکاسیون (fundoplication) لازم است. بیرونزدگی بیش از حد زبان باعث ترشح آب دهان میشود. درمانهای جراحی یا دارویی موجود (کشت مجدد مجاری بزاقی یا استفاده از چسبهای موضعی اسکوپولامین (scopolamine)) بهطور معمول موثر نیستند.
بسیاری از داروهای ضدصرع برای درمان تشنج در افراد مبتلا به سندرم انجلمن میشود؛ اما هیچ توافقی در مورد داروی بهینهی تشنج وجود ندارد؛ اگرچه والپروئیک اسید (valproic acid)، توپیرامات (topiramate)، لاموتریژین (lamotrigine)، لوتیراستام (levetiracetam) و کلونازپام (clonazepam) بیشتر در آمریکای شمالی استفاده میشود. استفادهی منفرد از دارو ترجیح داده میشود؛ اما پیشرفت تشنج رایج است. برخی از کودکان مبتلا به تشنج غیرقابل کنترل تحت رژیم کتوژنیک قرار گرفتهاند و این ممکن است در برخی موارد مفید باشد. گاهی از تحریم عصب واگ استفاده شدهاست.
اکثر کودکان مبتلا به Angelman syndrome برای رفتارهای بیشفعالی دارودرمانی دریافت نمیکنند؛ اگرچه برخی ممکن است از داروهای محرک مانند متیل فنیدات (methyl phenidate) (ریتالین) سود ببرند. اصلاح رفتاری در درمان رفتارهای نامطلوب که مخل اجتماعی یا خودآزاری هستند، موثر است. طیف کاملی از برنامههای آموزشی و غنیسازی باید در دسترس باشد. کودکان ناپایدار یا سراپایی ممکن است از فیزیوتراپی بهرهمند شوند. کاردرمانی ممکن است به بهبود کنترل حرکتی و دهانی-حرکتی کمک کند. صندلیهای تطبیقی یا پوزیشنرهای (positioners) مخصوص، بهویژه برای کودکان که دارای آتاکسی شدید هستند، ممکن است مورد نیاز باشد.
گفتاردرمانی برای بیماران مبتلا به سندرم انجلمن ضروری است و باید بر روشهای ارتباط غیرکلامی تمرکز کند. وسایل کمک ارتباطی مانند کارت تصویر یا تابلوهای ارتباطی باید در اولین زمان مناسب استفاده شوند. آموزش امضا باید به محض اینکه کودک به اندازهی کافی توجه کرد، آغاز شود. مشکلات ارتوپدی بهویژه “subluxed” یا مچ پاهای پرونیشن (pronated) یا تنگی تاندون آشیل را میتوان با جراحی اصلاح کرد.
در دوران کودکی ممکن است چاقی رخ دهد. رفتارهای مرتبط با غذا (خوردن اقلام غیرخوراکی، افزایش آشکار اشتها، افزایش جهتگیری رفتاری به غذا) در Angelman syndrome رایج است و میتواند در شروع چاقی نقش داشته باشد. مقداری افزایش وزن ممکن است در دوران جوانی رخ دهد. چاقی شدید در سندرم انجلمن بسیار نادر است. افراد مسنتر تمایل به تحرک و فعالیت کمتری دارند. توجه به برنامههای فعالیت میتواند مفید باشد و به کاهش چاقی کمک کند.
مشاوره ژنتیک
خطر ابتلا به خواهر و برادر یک فرد مبتلا به سندرم انجلمن به مکانیسم ژنتیکی سندرم انجلمن در پروباند (proband) بستگی دارد. پروباند به این معناست که شخص اصلی که اختلال روحی یا جسمی دارد، بهعنوان پایهی مطالعات توارثی یا ژنتیکی از او استفاده میشود.
برای مادرانی که دارای یک فرزند مبتلا به سندرم انجلمن با حذف بزرگ هستند، خطر ابتلا به Angelman syndrome در فرزندان آینده کمتر از 1 درصد است. این رقم 1% این واقعیت را در بر میگیرد که موزائیسم ژرملاین (germline mosaicism) برای حذف بزرگ در یک مورد گزارش شدهاست. همچنین در موارد نادر، مادر یک کودک سندرم انجلمن دارای یک جابهجایی کروموزومی متعادل است که مستعد عود سندرم انجلمن است. بنابراین به مادران کودکان مبتلا به سندرم انجلمن میتوان پیشنهاد کرد که آزمایش کروموزوم خون و FISH را برای بررسی بازآرایی مجدد کروموزوم 15 انجام دهند.
اگر بازآرایی کروموزومی یا حذف ناحیهی ژنی کوچک در پروباند شناسایی شده باشد، خطرات برای خواهر و برادر و سایر اعضای خانواده به ارثیبودن یا “de novo” بازآرایی بستگی دارد.
تشخیص قبل از تولد سندرم انجلمن
تشخیص قبل تولد از طریق شناسایی تمام تغییرات ژنتیکی مولکولی در ناحیهی 15q11.2-q13 امکانپذیر است و این کار را با کمک تجزیهوتحلیل DNA یا کروموزومی و FISH سلولهای جنینی بهدستآمده با نمونهبرداری از پرزهای کوریونی (chronic villus sampling) (CVS) یا آمینوسنتز (amniocentesis) انجام میدهند.
تجزیهوتحلیل متیلاسیون DNA بر روی سلولهای بهدستآمده توسط CVS از نظر تئوری امکانپذیر است؛ اما تعداد کمی از آزمایشگاههای بالینی که آزمایشهای قبل از تولد را با استفاده از تجزیهوتحلیل متیلاسیون DNA انجام میدهند، استفاده از آمینوسیتها را به دلیل هیپومتیلاسیون (hypomethylation) مرتبط با سلولهای مشتقشده از جفت را ترجیح میدهند.
غربالگری آمینوسنتز معمول (بهطور مثال در سنین بالای مادر) ممکن است با ناهنجاریهایی مواجه شود که جنین را در معرض خطر ابتلا به سندرم انجلمن قرار دهد. اگر در مطالعات سیتوژنیک از CVS یا آمینوسنتز به حذف 15q11.2-q13 مشکوک باشد، مطالعات FISH یا آرایهی CGH برای تایید حذف نشان داده میشود. اگر «حذف» تایید شدهاست، مطالعات منشا والد را میتوان انجام داد تا مشخص شود که حذف از مادر (جنین دارای سندرم انجلمن است) یا از پدر (جنین دارای سندرم پرادر ویلی (Prader-Willi syndrome) است) گرفته شدهاست.
نویسنده: ریحانه ارفعی
ویراستار: حدیث پرهیزگاری
منابع:
1.https://www.ninds.nih.gov/health-information/disorders/angelman-syndrome
2.https://medlineplus.gov/genetics/condition/angelman-syndrome/
3.https://europepmc.org/article/NBK/nbk1144
4.https://www.sciencedirect.com/science/article/pii/S1098360021015665