فهرست مطالب
چکیده
طیف شرایط ناشی از عملکرد غیر طبیعی CFTR گسترده است – از فیبروز کیستیک “کلاسیک” (CF گرفته تا شرایط اندام های مجزا که به عنوان اختلالات مرتبط با CFTR نامیده می شوند. تعیین و اطمینان از تشخیص در اقلیت مهمی از بیماران می تواند یک چالش باشد زیرا تست عرق امری ابهام دار یا طبیعی است. تأثیر این تست بر بیمار (در مراحل مختلف زندگی آنها) می تواند بسیار مهم باشد زیرا این پتانسیل را دارد که منجر به تشخیص نادرست و درمان بیش از حد با بار روانی مرتبط شود.
آزمایش اختلاف پتانسیل بینی و اندازه گیری جریان روده ، اندازه گیری های فیزیولوژیکی عملکرد CFTR است و بنابراین می تواند اطلاعات تشخیصی مهمی را ارائه دهد. در این مقاله ، مروری بر آخرین تحولات در تشخیص CF صورت گرفته است ، و تشریح این روش در صورت عدم قطعیت تشخیص و برخی از مناطق عدم قطعیت است.
فیبروز کیستیک چیست؟
فیبروز کیستیک (CF) یک بیماری مغلوب اتوزومی است که در اثر جهش در هر دو آلل ژن CFTR ایجاد می شود که تنظیم کننده هدایت گیرنده سیستمی فیبروز را رمزگذاری می کند. CFTR مسئول تنظیم حمل و نقل کلرید ، همراه با انتقال سدیم و آب در غشای آپیکال سلولهای اپیتلیال است. برای اکثر افراد مبتلا به CF ، تشخیص مستقیم و با شناسایی دو جهش ایجاد کننده بیماری در CFTR و / یا تست عرق تشخیصی (60 میلی مول در لیتر) در فرد با فنوتیپ سازگار تأیید می شود ( به عنوان مثال بیماری سینوپلمونری و نارسایی لوزالمعده یا از طریق غربالگری در نوزادان. با این وجود یک اقلیت مهم (تقریباً 10٪) وجود دارد که در آن تشخیص صحیح نیست ، زیرا تست عرق امری مبهم است (30-59 mol / l) یا حتی طبیعی (<30 میلی مول در لیتر) و دو جهش ایجاد کننده بیماری مشخص نشده اند.
این افراد اغلب می توانند سالها – یا حتی چند دهه – در سیستم های مراقبت های بهداشتی مبارزه کنند. یک برچسب تشخیصی ایمن ، که منجر به تشخیص غلط (به عنوان مثال “آسم”) یا برچسب های غیر مفید مانند “CF ممکن” یا “CF خفیف” می شود. این موارد دارای کاربردهای نامناسب بالینی ، روان شناختی و حتی مالی (به عنوان مثال بیمه عمر) برای بیمار به دلیل استفاده نامناسب از خدمات درمانی است. علاوه بر این ، در دوره جدید روشهای تعدیل کننده جهش CFTR، تأیید تشخیص با یک ژنوتیپ دقیق هرگز از اهمیت خاصی برخوردار نبوده است ، زیرا این روش های درمانی احتمالاً در طولانی مدت اصلاح کننده بیماری هستند.
ژنتیک
و دیگر مناطق MRNA سیگنالهای اتصال CFTR بیش از 1600 جهش در زنجیره کدگذار توصیف شده است.
جهش ها به طورکلی به 6 دسته طبقه بندی می شود که بر این اساس باعث بوجود آمدن بیماری می شود(جدول شماره 1) سه کلاس اول باعث افزایش تقرات در فنوتیپ افراد می شوند.
و توسعه ثانویه به کدون های پایانیCFTRکلاس 1 جهش ها باعث عدم وجود ترکیب ژن زودهنگام و یا سایر جهش های چارچوبی می شود.
معمولا در جهش های کلاس 2 رونویسی و ترجمه می شود.CFTR اما لایه های پروتئینی به طور نادرستی در طول انتقال های درون سلولی درشبکه اندوپلاسمی صاف شناخته می شود و به عنوان نقص معرفی می گردد. پروتئین قبل از رسیدن به محل سلولهای گیرنده در سطح سلول تخریب می شود.
جهش درجه 2(حذف یک باقیمانده فنیل آلانین) شایعترین جهش ژنتیکی بوده و حدود70 تا 73 درصد از آلل های معیوب را تشکیل می دهد.
در جهش کلاسه یک پروتئین به صورت نرمال در سطح سلول می باشد اما تنظیم ورود و یا انتقال دهنده کلرید باعث توقف فعالیت کانال یونی می شود. اگرچه جهش های درجه 3 دارای کمترین مقدار انتقال وابسته هستند.
نقص های کلاس 4 ناهنجاریهای را از رسانایی کلراید نشان می دهد چنین جهش هایی ممکن است باعث بوجود آمدن بیماری فیبروزکیستیک شود.
CFTRجهش های کلاس 5 تعداد رونوشت ها را کاهش می دهد و در نتیجه کانالهای عملکرد کمتری در سطح سلول پیدا می کند.
در نهایت جهش های کلاس 6 نقایصی را در پایداری پروتئین ایجاد می کند که باعث شتاب بخشیدن به گردش خون در سطح سلول می شود.
کلاسیک CF
بیماران فیبروز کیستیک کلاسیک دچار جهش های کلاس 1 و3 می باشند که باعث ایجاد بیماری های تنفسی اگزو کرین پانکراتیک می شوند که جهش های ژنتیکی خاص در آن به وجود نمی آید .
به عنوان مثال شایع ترین جهش از هموزیگوت بیش از حد بیماری را پیش بینی نمی کند اما این ژنوتیپ به عنوان عامل خطر مستقل در سایر فیبروزکیستیک ها از جمله کاه چگالی استخوان ها و…. نشان داده شده است.
تشخیص این نوع زمانی انجام می شود که بیماران حداقل یک ویژگی فنوتیپ خود را داشته باشند و این تشخیص می تواند به کمک یک تاریخچه خانوادگی و یا غربالگری های انجام شده صورت پذیرد .
علاوه بر این ها بیماران باید تست مثبتی با غلظت پتاسیم >60 میلی مول بر لیتر داشته باشد.آزمایش های ژنتیک نشان دهنده دو فیبروزکیستیک که باعث جهش ویا اثبات یک نشانه آزمایش فرعی غیرعادی می شوند مثل انتقال یونی اپی تلیال بینی یا اندازه گیری جریان روده .
چه کسانی مبتلا به این بیماری می شوند؟
این بیماری می تواند در ھر سنی فرد را گرفتار کند ولی رایج ترین سن ابتلا به بیماری در دھه پنجم است. زن و مرد به یک نسبت به آن گرفتار می شوند. اگر چه علت این بیماری روشن نیست ولی در ھر حال این یک عارضه عفونی نیز نمی باشد که از یک فرد به فرد دیگری سرایت کند و ھمچنین این عارضه شکلی از سرطان ھم نیست. برخورد با بعضی گرد و غبارھای شغلی(مثلا آزبست و یا ذرات فلزات سنگین) می تواند بیماری مشابھی را ایجاد کند. اکثر مبتلایان به این بیماری سیگاری بوده و یا در حال حاضر سیگار مصرف می کنند، اگر چه در بیشتر بیماران علت بیماری مشخص نیست.
عوارض فیبروز کیستیک چیست؟
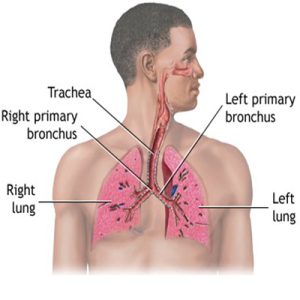
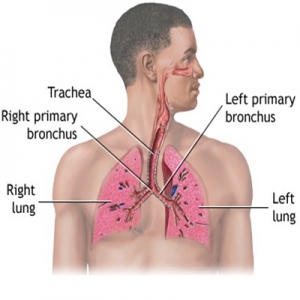
عوارض سیستم تنفسی در فیبروز کیستیک شامل موارد زیر میشود:
برونشکتاز، عفونتهای مزمن منجر به ذات الریه، رشد (پولیپ بینی)، هموپتیزی، پنوموتوراکس و در نهایت نارسایی تنفسی است.
از دیگر عوارض این بیماری می توان به ؛
نازایی، پوکی استخوان، عدم تعادل الکترولیتی و کم آبی بدن اشاره کرد که باعث افزایش ضربان قلب، خستگی، ضعف و فشار خون پایین می شود. به دلیل مداخلات پزشکی با کیفیت بهتر و مراقبت جامع، افزایش چشمگیر در درصد بیماران (29.2٪ در سال 1986 به 49.7٪ در سال 2013) که بالای 18 سال زنده مانده اند وجود دارد.
بیش از 7000 نفر فیبروز کیستیک در انگلستان دارند. این بیماری شایع ترین بیماری ارثی ژنتیکی در جمعیت های سفید پوست است (1 در 2500 نوزاد)، اگرچه به طور فزاینده ای در جمعیت های غیر سفید پوست مهم شناخته می شود.
با این وجود، بیشتر پزشکان عمومی فقط یک یا دو بیمار را در لیست خود دارند و از آنجا که مدیریت به طور کلی در مراکز تخصصی انجام می شود، بسیاری از پزشکان متخصص کودکان در مراقبت از تنها تعداد کمی از بیماران درگیر خواهند شد.
پیشرفت در درک ما از بیماری و تأثیر این بر مدیریت سریع بوده است
20سال گذشته فیبروز کیستیک به عنوان یک دستگاه گوارش و بیماری ریه در کودکان کوچک اما اخیراً نیز دیده شده است تبدیل به یک بیماری پیچیده و چندرسانه ای می شود که به آن گسترش می یابد.
بزرگسالی و فیبروز کیستیک
به زودی کودکان بزرگسالی با این بیماری وجود خواهند داشت. بقای متوسط پیش بینی شده برای نوزادان متولد قرن بیست و یکم اکنون بیش از 50 سال است. این افزایش بقا – همراه با تغییر در درمان استاندارد، اجرای بیشتر غربالگری نوزادان و تمرکز بر روی راهکارهای درمانی جدید – ما را به این فکر می اندازد که ممکن است بروزرسانی در مورد این بیماری بسیار نسبتاً نادر باشد
روش ها و الگوریتم های تشخیصی فیبروز کیستیک
در طول ده ها سال ، الگوریتم های تشخیصی مختلفی ایجاد شده اند ، اما اخیراً همکاری بین مقامات اصلی در آمریکای شمالی و اروپا باعث شده است که الگوریتم های قابل توجهی ایجاد شود ، و نتیجه آن توافق برای آستانه های تشخیصی کلرید عرق (یعنی دامنه دو برابر در کلرید عرق) است. حرکت از 40-59 میلی مول در لیتر به 30-59 میلی مول در لیتر) و آزمایش فیزیولوژیکی CFTR (اندازه گیری اختلاف پتانسیل بینی [NPD] و اندازه گیری جریان روده [ICM]) که جزء لاینفک کار در بیماران مبتلا به مبهم است.
هر دو NPD و ICM حمل و نقل یونی تنظیم شده با CFTR را نشان می دهند – منعکس کننده کلرید خالص و حرکت سدیم در غشای آپیکال سلولهای اپیتلیال – اما NPD یک ارزیابی داخل بدن از دستگاه تنفسی (در بینی) و ICM یک ارزیابی داخل بدن از بافت رکتوم است. هر دو توانایی ارائه اطلاعات اضافی مهم در مورد عملکرد CFTR را دارند و بنابراین می توانند عملکرد تشخیصی را افزایش دهند.
پروتکل های استاندارد برای هر دو روش موجود است ، اگرچه استفاده از NPD گسترده تر است ، از جمله در خدمات تشخیصی CF دشوار بیمارستان خصوصی برومپتون. داده ها برای NPD نشان داده است که در مقایسه با آزمایش عرق ، بیماران بیشتری را به عنوان داشتن CF با داشتن نتایج کمتر از مرز در یک گروه از بیماران مبتلا به بیماری تک اندام (33.2 ab غیر طبیعی برای NPD در مقابل 17.3 for برای آزمایش عرق) شناسایی کرده است.
مهمتر اینکه ، تجزیه و تحلیل ژنتیکی گسترده CFTR کمترین حساسیت بود و تنها با افزایش 8/8٪ عملکرد تشخیصی ، این شامل افزایش 122 جهش به عنوان بیماری ایجاد شده توسط CFTR2 ، مخزن پایگاه داده برای اطلاعات ژنوتیپ CFTR شد. با این وجود ، توجه به این نکته مهم است که از این انتشار ، تعداد جهش های ایجاد کننده بیماری تعریف شده توسط CFTR2 به 312 افزایش یافته است. ICM نیز با روشی مشابه مورد مطالعه قرار گرفته است که نشان دهنده پتانسیل تبعیض آمیز برای بیماران مبتلا به تست های عرق دو قطبی است. همچنين اين امكان را دارد كه در كودكان خردسال امكان پذير باشد (تا زماني كه آنها بتوانند بيوپسي را تحمل كنند) و در بيماران مبتلا، پليپوز بيني يا التهاب قابل توجهي دارند.
ژنوتیپ فیبروز کیستیک
یک مسئله پیچیده دیگر در مورد تشخیص CF ، به ویژه در زمینه بیمار “غیر معمولی” ، ژنوتیپ است ، زیرا با وجود CF یک بیماری تک زا ، دو نوع ممکن است در یک فرد شناسایی نشوند و حتی اگر وجود داشته باشد ، اهمیت بالینی یک یا هر دو نوع ممکن است ناشناخته باشند. بیشتر آزمایش های کیت ژنوتیپ CFTR در دسترس تجاری بین 36 و 50 جهش فراهم می کند که پوشش آلل 85-85٪ برای جمعیت داده شده را فراهم می کند.
بنابراین ، اگر بیمار از اقلیت قومی باشد ، این کیت برای فرد حساسیت کمتری خواهد داشت. حتی با تجزیه و تحلیل CFTR گسترده ، اکثر سیستم عاملهای توالی بالینی کل ژن را دنباله نمی کنند ، پوشش اگزون ها ، مرزهای اگزون / اینترون را ارائه می دهند و گاهی اوقات انواع intronic را انتخاب می کنند.
در حال حاضر بیش از 2000 نوع مختلف در CFTR شناسایی شده اند ، اما در حال حاضر تنها نسبتاً کمی از این افراد عامل بیماری هستند. تاکنون 374 نوع آن به طور گسترده در CFTR2 مورد مطالعه قرار گرفته و 312 عامل بیماری در نظر گرفته شده است. از بین باقیمانده ، 36 مورد با عواقب بالینی متغیر همراه هستند ، 13 غیر CF-ایجاد کننده در نظر گرفته شده و برای 13 نوع دیگر ارزیابی بی نتیجه بوده است. بنابراین ، بیماران با هر نوع بیماری که بیماری محسوب نمی شوند
برای اثبات اختلال عملکرد CFTR باید به دقت ارزیابی شود ، زیرا تنها در صورت وجود این بیماری می توان تشخیص CF را در نظر گرفت. تقویت پروب چند منظوره (MLPA) نیز برای تشخیص حذف کامل اگزون یا تکثیر انجام می شود. به طور کلی ، حساسیت این روش در منطقه از 98-95٪ در نظر گرفته شده است ، بنابراین نشان می دهد که با وجود این فناوری ، هنوز هم می توان در موارد نادری انواع مختلفی از دست داد. در حال حاضر بیش از 2000 نوع مختلف در CFTR شناسایی شده اند ، اما در حال حاضر تنها نسبتاً کمی از این افراد عامل بیماری هستند.
تاکنون 374 نوع آن به طور گسترده در CFTR2 مورد مطالعه قرار گرفته و 312 عامل بیماری در نظر گرفته شده است. از بین باقیمانده ، 36 مورد با عواقب بالینی متغیر همراه هستند ، 13 غیر CF-ایجاد کننده در نظر گرفته شده و برای 13 نوع دیگر ارزیابی بی نتیجه بوده است. بنابراین ، بیماران با هر نوع واریزی که عامل بیماری محسوب نمی شوند ، برای اثبات اختلال عملکرد CFTR باید با دقت ارزیابی شوند ، زیرا تنها در صورت وجود این بیماری می توان تشخیص CF را در نظر گرفت.
با معرفی تعدیل کننده های CFTR ، تمرکز تازه ای در طبقه بندی جهش های CFTR صورت گرفته است. از نظر تاریخی ، آنها بر اساس تأثیر آنها بر سنتز پروتئین و عملکرد آنها ، به پنج یا شش گروه تقسیم شده اند. بیماران هموزیگوت یا هتروزیگوت مرکب برای جهش از کلاسهای I تا III معمولاً دارای نارسایی لوزالمعده اگزوکرین و مقدار کلرید عرق بالای 60 میلی مول در لیتر هستند ، در حالی که بیمارانی که حداقل یک کلاس جهش IV تا VI دارند معمولاً مشکل لوزالمعده دارند. با این حال ، مطالعات اخیر محدودیت های این سیستم را برجسته کرده است زیرا بسیاری از جهش ها با هم همپوشانی دارند.
در نتیجه ، مفهوم “عملکرد باقیمانده” یا “عملکرد حداقل” جهش به طور فزاینده ای مورد استفاده قرار می گیرد ، که در آن جهش هایی که شواهدی از شواهد بالینی و آزمایشگاهی از عملکرد CFTR باقیمانده را نشان می دهند (به عنوان مثال پروتئین بالغ CFTR بر روی وسترن بلات و شواهد آزمایشگاهی) وجود دارد. حمل و نقل کلرید) با هم گروه بندی می شوند ، زیرا پیش بینی می شود در مورد تعدیل کننده های خاص CFTR پاسخگو باشند. در حالی که پیش بینی نمی شود که جهش های عملکردی حداقل به تعدیل کننده های فعلی CFTR پاسخ دهند ، زیرا معمولاً پروتئین CFTR بالغ تولید نشده یا شواهد آزمایشگاهی انتقال کلرید وجود ندارد.
روش های درمانی فیبروز کیستیک
در حال حاضر ، اقدامات پیشگیرانه محدود به برنامه های بهداشت ریوی است که شامل فیزیوتراپی ، استنشاق dornase alfa و جلوگیری از استعمار متقاطع در بین بیماران است. رویکرد پیشگیری و درمان عفونت های ریه در بیماران مبتلا به CF در مرکز CF در کپنهاگ مبتنی بر کنترل ماهانه سفت و سخت است ، که شامل معاینه میکروبیولوژیکی ترشحات دستگاه تنفسی تحتانی است. هر زمان که پاتوژنها شناسایی شوند ، صرف نظر از شرایط بالینی بیمار ، درمان آغاز می شود
s. pneumoniae
این ارگانیسم در کودکان شایعتر از بزرگسالان در جمعیت های دارای و بدون CF است. همانطور که در جمعیت غیر CF ، میزان ریشه کنی آن پس از 2 هفته انجام مونوتراپی دهان نزدیک به 100٪ است.
H. آنفلوانزا
این ارگانیسم علی رغم شیوع بالای آن در ایزوله های بالینی ، به ندرت در CF مداوم یا مزمن است. درمان شده با 2 هفته آنتی بیوتیک خوراکی به عنوان تک درمانی ، میزان ریشه کنی آن در حدود 70٪ 0.8.9 است.
aureus در CF ، میزان ابتلا به عفونت ریه با این پاتوژن به طور غیرمعمول زیاد است ، و خطر قابل ملاحظه ای از مزمن بودن آن وجود دارد. بنابراین ، مطلوب است که اثرات درمانی را با استفاده از تایپ فاژ و آنتی بادی های سرم خاص کنترل کنیم.
درمان ترکیبی با آنتی بیوتیکهای خوراکی با دوزهای بالا به مدت 2 هفته منجر به ریشه کنی در حدود 70٪ می شود. بروز اولین جداسازی P. aeruginosa در CF با انتقال مستقیم بیمار به بیمار ، 10،11 افزایش می یابد و می تواند کاهش یابد. توسط جداسازی کوهورت.12 محاکمه واکسن های مختلف P. aeruginosa معمولاً نتایج دلسرد کننده داشته است.
علیرغم میزان اولین بار کسب این ارگانیسم که می تواند به اندازه 1 تا 2٪ در سال باشد ، 12 شکل 1 نشان می دهد که شیوع استعمار بسیار زیاد است .6 این قابل توضیح است زیرا ، اگر درمان نشود ، استعمار اولیه به بسته به میزان قرار گرفتن در معرض عفونت مزمن غیر قابل برگشت در حدود 12 ماه.
در این مرحله اولیه علائم عفونت باکتریایی در دستگاه تنفسی تحتانی (LRT) وجود ندارد. بنابراین ، توسعه عفونت مزمن P. aeruginosa ممکن است به عنوان یک زنجیره ای توصیف شود که با افزایش تدریجی تعداد موجودات موجود در درخت برونش مشخص می شود. با این وجود شواهدی وجود دارد که حاکی از عدم بازگشت پس از آن ارگانیسم ها از LRT نیستند.
به نظر می رسد این نقطه با دو رویداد مرتبط است: ظهور آنتی بادی های سرم خاص قابل تشخیص در برابر P. aeruginosa14 و فعال سازی ژنهای P. aeruginosa که کد تولید آلژینات است. حالت دوم مکانیسم بقا است که به ارگانیسم ها می تواند میکروکلون هایی را که در یک بیوفیلم آلژینات تعبیه شده است ، ایجاد کند و آنها را با مکانیسم های دفاعی میزبان یا داروهای ضد میکروبی مستعدتر می کند.
این احتمال وجود دارد که این دو واقعه ، از نزدیک با هم مرتبط باشند ، به صورت علنی نیز مرتبط هستند ، اما جزئیات هنوز مشخص نیست. افزایش در سطح اختصاصی آنتی بادی ممکن است افزایش بار آنتی ژنی به دلیل رشد تکثیر میکروکلونهای P. aeruginosa باشد. از طرف دیگر ، تعامل آنتی بادی های خاص با آنتی ژن های P. aeruginosa ممکن است مکمل جذب و فعال سازی PMN ها را فعال کند. گونه های بسیار واکنشی از اکسیژن مولکولی تولید شده در طول انفجار اکسیداتیو PMN ها ممکن است ژن های P. aeruginosa را که برای تولید آلژینات رمزگذاری شده اند ، فعال کنند.
جالب است که ، آنتی بیوتیک های ماکرولید تولید آلژینات را در شرایط in vitro سرکوب می کنند ، و از این طریق ممکن است ارگانیسم ها از یک مکانیسم مهم بقا محروم شوند. به تازگی ، نشان داده شده است که درمان آزیترومایسین باعث افزایش ظرفیت اجباری حیاتی (FVC) و حجم منقضی شده اجباری در 1 ثانیه (FEV1) در بیماران مبتلا به عفونت مزمن P. aeruginosa می شود ، و در نتیجه ممکن است جایی برای مداوم آزیترومایسین در این بیماران وجود داشته باشد.
نقش احتمالی سرکوب آلژینات در درمان اولیه استعمار متناوب P. aeruginosa و عفونت نیاز به بررسی دارد. در مطالعه دستیابی به عفونت مزمن P. aeruginosa در کلینیک ما ، از یک تعریف دقیق استفاده می شود که در مورد نظارت روزانه میکروبیولوژیکی ماهانه بیماران مبتلا به CF پیش بینی می شود. ما در نظر می گیریم که عفونت مزمن یا هنگامی که P. aeruginosa از ترشحات LRT جدا شده باشد (بدست آمده از طریق بیرون کشیدن یا مکش بینی) در هر یک از شش بازدید مداوم ماهانه انجام شود.
یک مطالعه گذشته نگر نشان داد که شیوع عفونت مزمن در طی سالهای حضور کلینیک ما متفاوت بوده است. اینها نکات زمانی برجسته هستند: درمان ضد پلاسمی داخل وریدی انتخابی هر 3 ماه در بیماران مبتلا به عفونت مزمن در سال 1975 در کلینیک جداگانه آغاز شد و بخش های جداگانه در بیماران مبتلا به CF با فرهنگ مثبت و منفی P. aeruginosa در سال 1980 آغاز شد و یک کارآزمایی بالینی آینده نگر از درمان تهاجمی ضد پوستی استعمار اولیه در سال 1988 آغاز شد.
30سال پیش این بیماری به طور مداوم منجر به مرگ در دهه اول زندگی و گاهی وقتها نیز باعث وخامت ریه و ورود باکتری های فرصت طلب و در نهایت باعث شدید تر شدن بیماری می شود .
پیشرفت ها در درمان این بیماری در نهایت باعث بهبود قابل توجهی در بقاء و طول عمر این بیماران تا 36 الی 38 سال شد.
بر اساس ژنتیک این بیماری . بیماری مربوط به اختلال یا نقص تنظیم کننده رسانای غشاء است که یک آنیون راسی غشاء می باشد (کلراید و بی کربنات) که کانال در بافت پوششی تنفسی و اگزوکرین ها می باشد.
تشخیص های اولیه فیبروزکیستیک برای اجازه و مداخله های قبل از اینکه بیماری ریوی پیشرفت کند حیاتی و لازم است.
شاخص های کیفیت زندگی نشان داده است که بیماران سینوزیت اغلب دارای عملکرد های ریوی هستند و می تواند پیش بینی کننده بیماران ریوی به خصوص در کودکان باشد.
علاوه بر این ها چند آزمایش کنترل شده تصادفی در حال حاضر با توجه به کارایی روش های درمان فیبروز کیستیک در دسترس هستند و بسیاری از مطالعات به دلیل عدم پیگیری های طولانی مدت محدود شده اند. در واقع دستورالعمل های مبتنی بر شواهد ناقص هستند و الگوهای مدیریتی مربوط به مداخلات بیماری برای همه ی بیماران استاندارد نشده است.
نتیجه گیری
تشخیص دقیق CF برای بیماران ، خانواده ها و خدمات درمانی بسیار ضروری است. بدون آن ممکن است افراد از اهمیت بالقوه مراقبت در حال تغییر زندگی برخوردار نباشند ، یا برعکس اگر تشخیص صحیح CF نباشد ، ممکن است تحت معالجه قرار گیرند. درک ما از طیف CF و نتایج متغیر آن طی چند دهه به طرز چشمگیری افزایش یافته است ، اما در عصر توالی ژن، ژن های بعدی هنوز چیزهای زیادی برای آموختن وجود دارند.
در حالی که ما پتانسیل خوبی برای شناسایی انواع نادر CFTR داریم ، درک کامل عواقب عملکردی اینها از طریق آزمایش دقیق فیزیولوژیکی برای درک بهتر پیامدهای بالینی بسیار مهم است. در حال حاضر ، هیچ آزمایش تشخیصی CF وجود ندارد که به اندازه کافی دقیق باشد تا در انزوا مورد استفاده قرار گیرد.
دستورالعمل های آینده شامل استفاده از به اصطلاح “ارگانوئیدها” یا “اندامهای کوچک” است که از سلولهای بنیادی بیماران جدا می شوند تا عملکرد CFTR جهش های فردی خود را ارزیابی کنند – این یک نمونه هیجان انگیز از پزشکی شخصی است ، اما آیا این ارتباط با فرد ارتباط دارد یا خیر. تظاهرات بالینی خاص و خطر در طولانی مدت ، یک سؤال مهم تحقیق است.
درنهایت ، بهترین استراتژی فعلی برای تشخیص ، رویکردی چند وجهی است ، که یکپارچه سازی ارزیابی بالینی دقیق با ژنوتیپ و آزمایش عملکردی کامل CFTR ، از جمله NPD و / یا ICM ، هنگامی که تست های استاندارد دوقطبی است. از آنجا که عواقب طولانی مدت بیماران مبتلا به این ویژگی ها به خوبی درک نشده است ، مهم است که هر زمان داده های ممکن به صورت طولی جمع آوری شود تا دانش خود را به ما اطلاع دهند و در نهایت اطلاعات دقیق تری را برای بیماران خود در طول زندگی خود ارائه دهیم.
نویسندگان: نیلوفرممبینی، مهسا نیکجو، زهرا رضائی، بهشاد والی زاده، فرشته خلیل زاده، مهدیه بازدار، یاسمین نوابی
منابع:
- Koch C. Early infection and progression of cystic fibrosis lung disease. Pediatric pulmonology. 2002;34(3):232-6.
- Chaaban MR, Kejner A, Rowe SM, Woodworth BA. Cystic fibrosis chronic rhinosinusitis: a comprehensive review. American journal of rhinology & allergy. 2013;27(5):387-95.
- Simmonds N. Is it cystic fibrosis? The challenges of diagnosing cystic fibrosis. Paediatric respiratory reviews. 2019.
- 4. Rafeeq MM, Murad HAS. Cystic fibrosis: current therapeutic targets and future approaches. Journal of translational medicine. 2017;15(1):84.
- Davis PB. Cystic fibrosis since 1938. American journal of respiratory and critical care medicine. 2006;173(5):475-82.
همچنین بخوانید:سندروم XGS
