فهرست مطالب
مقدمه
تنظیم طول تلومرها در سلولهای بنیادی پرتوان[1] ارگانیسم های چندسلولی برای اطمینان از رشد و بقای ارگانیسم بحرانی است. تلومرها از DNAی تکرارشونده تشکیل شده اند که با هر تقسیم سلولی رفته رفته از بین میروند. وقتی تلومرها بشدت کوتاه شوند، پاسخ آسیب به DNA را راه میاندازند که به توقف چرخه ی سلولی می انجامد. سلول های بنیادی پرتوان درجهت بی اثرکردن سایش تلومر به مکانیسم های کشیدگی تلومر تجهیز شده اند که ظرفیت تکثیر و خودسازی طولانی مدت را تضمین میکنند.
کشیدگی بیش ازحد تلومر مخرب است و با مکانیسم حذف سریع تلومر بی اثر میشود که پیرایش تلومر[2] نامیده میشود. درحالیکه پیامدهای تلومر بشدت کوتاه کاملاً محزر است، ما در آغاز مسیر درک مکانیسم های خنثی کنندگی بیش ازحد تلومر هستیم . توازن بین کوتاه شدگی و کشیدگی تلومر ، نقطه ی تنظیم طول تلومر را در سلولهای بنیادی پرتوان تعیین میکند و پتانسیل تکثیر پایا را بدون ناپایداری کروموزومی تضمین میکند.
کلمات کلیدی: تلومر، سلولهای بنیادی، شلترین[3]، TZAP ، ZBTB48، پیرایش تلومر.
چگونه تلومرها توسط سلول های بنیادی کنترل می شوند؟
1- تنظیم طول تلومر
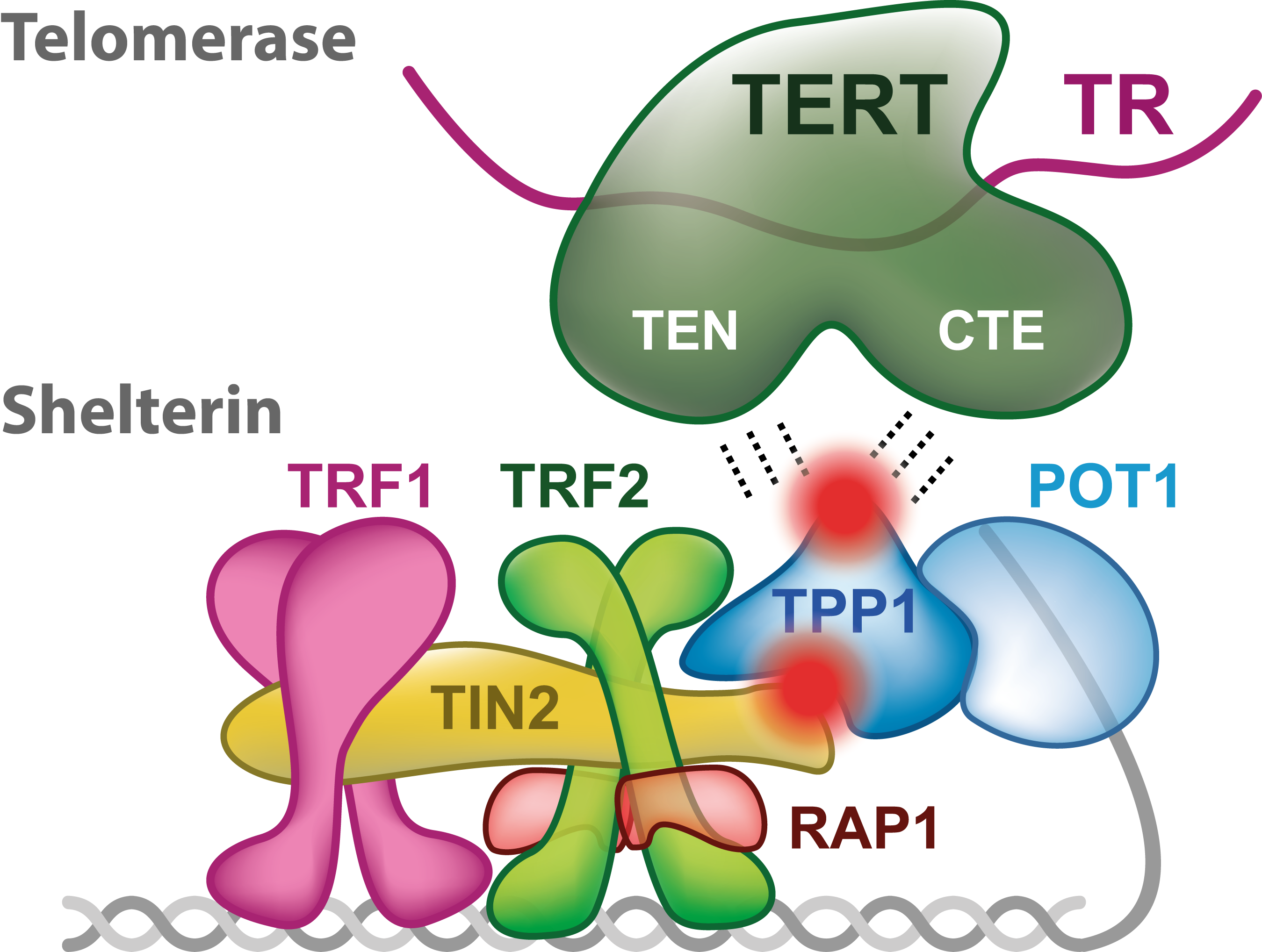
تلومرها ساختارهای نوکلئوپروتئینی ضروری هستند که برای پوشش و حفاظت پایانه های کروموزومی لازم هستند. تلومرها در پستانداران از توالی های تکرارشونده ی [TTAGGG]n تشکیل شده اند که سایت اتصال کمپلکس پروتئینی حفاظتی بنام شلترین هستند (de Lange, 2005). شلترین، کمپلکس شش پروتئینی متشکل از دو پروتئین اتصالی دورشته ای بنام TRF1 و TRF2 است که باقیمانده ی کمپلکس (TIN2-TPP1-POT1 و RAP1) را بطور اختصاصی از پایانه های کروموزومی جذب می کند.
کمپلکس شلترین DNAی تلومری را داخل حلقه ی تی[4] ثانویه طناب مانند شکل می دهد که از اشغال (تهاجم) بیرون زدگی تلومری 3′ تک رشته ای درون ناحیه ی تلومری دو رشته ای حاصل میشود (Doksani و همکاران، 2013؛ Griffith و همکاران، 1999). حفاظت پایانه ی کروموزومی از تخریب نوکلئولیتیک و فعالسازی پاسخ آسیب به DNA توسط ساختارهای حلقه ی تی و نیز اتصال کمپلکس شلترین تضمین میشوند (Denchi، 2009؛ Denchi و de Lange، 2007؛ Doksani و همکاران، 2013؛ Karlseder و همکاران، 2004؛ Okamoto و همکاران، 2013؛ Sfeir و de Lange، 2012).
کوتاه شدگی تدریجی تلومر
کوتاه شدگی تدریجی تلومر با تقسیم سلولی بعلت ناتوانی پلیمرازهای DNA در همانندسازی کامل الگوی خطی رخ می دهد که اصطلاحاً مسئله ی همانندسازی نهایی نامیده میشود (Olovnikov، 1973؛ Watson، 1972). درنتیجه، همانندسازی سلول ها دستخوش سایش پیشرونده ی تلومر میشود که اگر توسط مکانیسم های کشیدگی تلومر بی اثر شود، به تلومر های بشدت کوتاه می انجامد که کمپلکس شلترین کافی را احیا نمیکنند.
طول تلومر ، تعیین کننده ی اصلی پتانسیل تکثیر در سلول های فاقد مکانیسم های کشیدگی تلومر مثل لاین های سلول سوماتیک انسان است. کشیدگی تلومر توسط تلومراز، آنزیم مرکب از ترانسکریپتاز معکوس TERT و الگوی RNA، TERC و نیز پروتئین های اتصالی همچون ریبونوکلئوپروتئین دیسکرین[5] تضمین می شود (Cohen و همکاران، 2007). عملکرد تلومر، افزودن مجدد تکرارهای TTAGGG به پایانه های کروموزومی با امکان بازپُرسازی توالی های پایانی از دست رفته بعلت مشکل همانندسازی پایانی است (Blackburn، 1997) (شکل یک). درنتیجه، کشیدگی تلومر در سلولهای زایا و بنیادی برای اطمینان از تقسیمات سلولی کافی برای رشد، تکثیر و تقسیم بافتی[6] و بازسازی بافت[7] بحرانی است.
بیان تلومراز در پستانداران دارای عمر طولانی مثل انسانها در اکثر بافت های سوماتیک بیان می شود (Gomes و همکاران، 2011). برخی بیماری های مرتبط با کشیدگی معیوب تلومر جمعا اختلالات بیولوژی تلومر (TBD) نامیده میشوند که اهمیت تنظیم درست طول تلومر را مشخص میکنند (Savage، 2014). تلومرهای بیماران درگیر این بیماری ها بشدت کوتاه هستند و علائم مرتبط با تکثیر سلولی معیوب بسته به شدت بیماری بروز می کنند (Savage، 2014).
فرایند مبتنی بر نوترکیبی بنام کشیدگی جایگزینی تلومرازها (ALT)
تلومر ها ازطریق فرایند مبتنی بر نوترکیبی بنام کشیدگی جایگزینی تلومرازها (ALT) می توانند به شیوه ی مستقل از تلومراز کشیده شوند (Bryan و همکاران، 1997) (شکل یک). کسر قابل توجهی از سلول های سرطانی (تقریباً 15 درصد) تلومراز را بیان نمیکنند و تلومر را با کمک مسیر ALT حفظ می کنند. ALT دستگاه نوترکیبی همولوگ را برای استفاده از توالی های تلومری بعنوان الگوی کشیدگی تلومر بکار می گیرد (Dunham و همکاران، 2000).
جالب اینجاست ALT در سلولهای سوماتیک موشی تبدیل نشده رخ میدهد (Neumann و همکاران، 2013). همچنین گزارش شده است که مکانیسم های شبه ALT درطی مراحل اولیه ی جنین زایی[8] فعال هستند (Liu و همکاران). در این موارد، جنبه های شبه ALT همزمان با بیان تلومراز رخ می دهند.
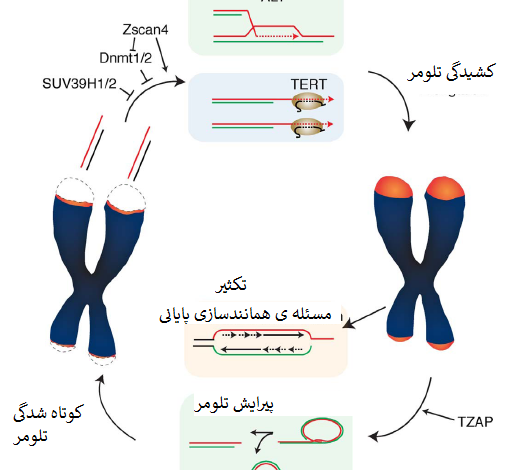
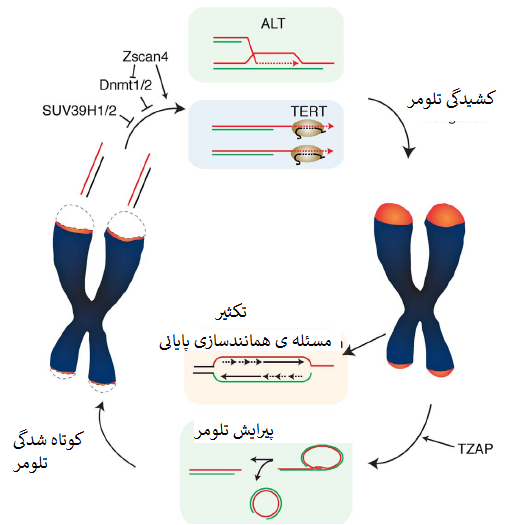
درحال حاضر مشخص نیست چه میزان از این مکانیسم های مستقل از تلومراز در کشیدگی تلومر در سلول های نرمال نقش دارند. کشیدگی تلومر با فرایندی بنام پیرایش تلومر متوازن میشود که طول تلومر را ازطریق حذف فعالانه ی تلومرهای فوق العاده طویل مضر در ثبات ژنوم بصورت وارونه تنظیم میکند (Pickett و همکاران، 2009) (شکل یک).
درحالی که مکانیسم های کشیدگی تلومر که کران پایین طول تلومر را حفظ میکنند بمدت چندین دهه کاملا بررسی شده اند، نحوهی تعیین حد بالایی طول تلومر ازطریق پیرایش تلومر بخوبی درک نشده است. دراینجا پیشرفت های اخیر در حوزه ی کنترل طول تلومر با تاکید ویژه روی توازن بین کشیدگی و پیرایش در سلول های بنیادی پرتوان ارائه میشوند.
2- کشیدگی تلومر در سلولهای بنیادی پرتوان
سلول های بنیادی پرتوان مثل سلول های بنیادی جنینی (ESC) قادر به خودسازی هستند و درواقع به هر نوع سلول سوماتیک منتهی می شوند. سلول های بنیادی جنینی، نخستین سلول های بنیادی پرتوان بودند که در آزمایشگاه جدا و کشت شدند (Evans و Kaufman، 1981؛ Martin، 1981). سلول های بنیادی جنینی موش در زمینه ی ژنتیک موش انقلابی بپا کردند بدین علت که نامیرا (جاویدان) هستند، به لحاظ ژنتیکی می توانند تغییر داده شوند و برای ایجاد هر موشی حتی پس از ماه ها کشت آزمایشگاهی استفاده شوند.
اخیراً ساخت کشت های سلول های بنیادی جنینی انسان علاوه بر قابلیت خلق سلول های بنیادی پرتوان القا شونده (iPSCs) از سلول های سوماتیک نویدبخش درمان های پیوندی جدید بوده اند (Takakshi و Yamanaka، 2006؛ Thomson و همکاران، 1998). ESCها و iPSCها می توانند تلومر ها را طویل کنند و درعین حفظ ثبات ژنومی و پرتوانی بی نهایت تکثیر شوند. مشخص کردن اهمیت همایستایی[9] تلومر در این سلول ها این یافته است که پرتوانی ESCها با تلومر های کوتاه کم شده است و دچار نقایص تمایزی هستند (Pucci و همکاران، 2013).
کشیدگی تلومر در ESCها
کشیدگی تلومر در ESCها با مکانیسم های چندگانه ازجمله سطوح افزایش یافته ی فعالیت تلومراز تضمین میشوند (Thomson و همکاران، 1998). جالب اینجاست که فعالیت شبه ALT در ESCها مشاهده شده است (Liu و همکاران، 2007). با این فرض که نقصان فعالیت تلومرازی ESCها به کوتاه شدگی شدید تلومر می انجامد (Huang و همکاران، 2011؛ Niida و همکاران، 1998؛ Pucci و همکاران، 2013)، نقش مکانیسم شبه ALT در هم ایستایی تلومر در ESCها باید مشخص شود.
جنبه ی متمایز ESCها ازطریق ساختار کروماتین تلومریک باز با سطوح کاهیده ی مارکرهای هتروکروماتین H3K9me3 و H4K20me3 ارائه شده است. نبود این مارکرها مطرح می کند که کروماتین تلومریک در ESCها ازهم باز شده است، وضعیتی که با افزایش کشیدگی توسط تلومراز مرتبط شده است. درواقع کاهش H3K9me3 بواسطه ی نقصان SUV39H1/H2 یا کاهش H4K20me3 بواسطه ی نقصان Suv4–20 h هردو کشیدگی تلومر را القا میکنند (Benetti و همکاران، 2007؛ Gomes و همکاران، 2011). بیان Zscan4 درطی رشد موش به کشیدگی تلومر از طریق کاهش متیل زایی DNA می انجامد (Dan و همکاران، 2017؛ Zalzman و همکاران، 2010).
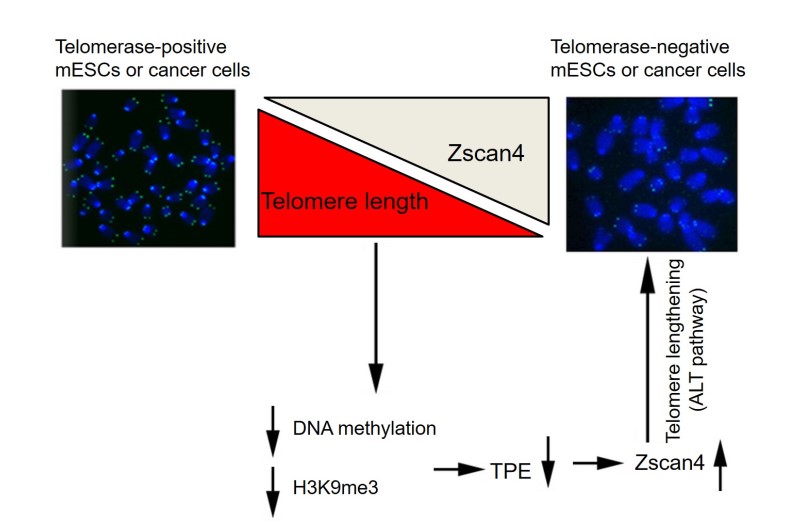
فقدان متیل ترانسفرازهای DNA و کشیدگی تلومر
فقدان متیل ترانسفرازهای DNA در سلولهای بنیادی جنینی موش (Dnmt1، Dnmt3a/3b) به کشیدگی تلومر می انجامد (Gonzalo و همکاران، 2006). این درحالی است که پیامد فقدان متیل ترانسفراز DNA از لحاظ کشیدگی تلومر در سلولهای انسان متفاوت است. بیماران فاقد DNMT3b تلومرهای بسیار کوتاه دارند و دچار نقص ایمنی، ناپایداری سانترومری و سندروم ناهنجاری های صورت (ICF) هستند (Yehezkel و همکاران، 2008). این ناهمخوانی میتواند بعلت تفاوت های ویژه بین سلولهای انسان و موش ازلحاظ تنظیم طول تلومر یا فاکتورهای تغییردهنده ی اضافی باشد که در کوتاه شدگی تلومر در بیماران ICF نقش دارند.
جالب اینجاست که سطوح متیل زایی کاهیده همچنین با فعالیت های شبه ALT همچون افزایش تبادل کروماتید خواهری تلومر (TSCE) مرتبط شده اند (Dan و همکاران، 2017؛ Zalzman و همکاران، 2010). سطوح پایین H3K9me3 و H4K20me3 در سلول های سرطانی گزارش شده اند که از مسیر ALT برای کشیدگی تلومرها استفاده میکنند (Episkopou و همکاران، 2014).
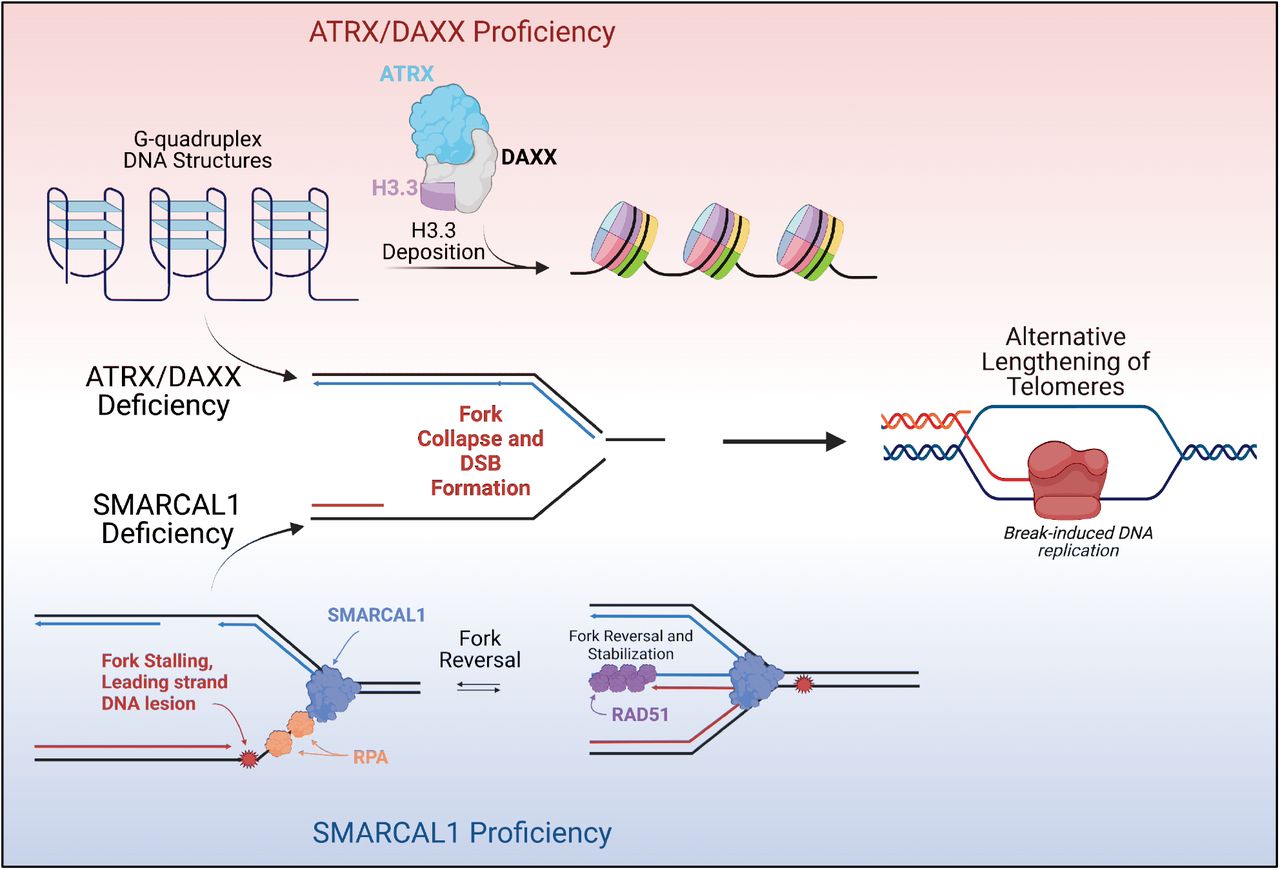
ALT در سلول های سرطانی با جهش ها در کمپلکس باز آرایی ATRX/DAXX و واریته ی هیستونی H3.3 همبسته است (Heaphy و همکاران، 2011؛ Lovejoy و همکاران، 2012). کمپلکس ATRX/DAXX بعنوان یک چاپرون عمل می کند که هیستون H3.3 را در هتروکروماتین پریسنتریک، تلومرها، و نیز سایتهای هتروکروماتیک درکل ژنوم ته نشین می کند (Voon و همکاران، 2015). این داده ها درمجموع ارتباط بدیهی بین وضعیت کروماتین و مکانیسم های کشیدگی تلومر را نشان می دهند، موضوعی که در بررسی های قبلی بطور گسترده پوشش داده شده است (برای جزئیات بیشتر مشاهده کنید OSullivan و Almouzni، 2014).
3- شواهد آستانه ی بالای طول تلومر در مخمر
درحالی که نیاز به حفظ آستانه ی پایین طول تلومر کاملا محرز شده است، مشخص نیست آیا آستانه ی بالایی طول تلومر در حفظ ثبات ژنومی مهم است یا خیر. نشانه های اولیه ی کران بالایی طول تلومر از انالیز سویه ی ساکارومایسس سرویزیه[10] حامل آلل های جهش یافته ی Rap1 نشات می گیرد (Kyrion و همکاران، 1992). تلومر ها در این سویه ی جهش یافته تا 4 kb از اندازه ی نرمال تقریباً 300 bp طویل شده اند (Kyrion و همکاران، 1992؛ Lustig و Petes، 1986). این تلومر های طویل شده به سبب نرخ های افزایش یافته ی فقدان کروموزوم بسیار ناپایدار بودند.
انالیز سرنوشت سلول های حامل این تلومر های اَبَرکشیده، فرایندی بنام حذف سریع تلومر (TRD) را آشکارکرد که می تواند تلومر های فوق العاده ی طویل بازگشتی به طول WT را بازنشانی کند. حذف سریع تلومر ازطریق مسیر نوترکیبی همولوگ رخ داد که به لحاظ مکانیکی از فقدان تدریجی تلومرها، بنام سایش تلومری، مجزا است (Li و Lustig، 1996). TRD به لحاظ ژنتیکی به فاکتورهای نوترکیبی همولوگ Mre11 و Rad50 نیاز دارد.
نقصان تلومر مرتبط با پروتئین Taz1, کشیدگی زیاد تلومر و نقایص همانندسازی تلومر
بعدها مشخص شد که سلول های میوزی در ساکارومایسس سرویزیه برای حفظ اندازه ی تلومری نوع وحشی در فرایند های که شبیه TRD است، دستخوش نرخ های بالای حذف دقیق میشوند (Joseph و همکاران، 2005). TRDها در این بررسی 30 تا 70 برابر بزرگتر از سلول های میوزی بودند که مطرح می کند.
کنترل کران بالایی طول تلومر مخصوصا در این مرحله از رشد اهمیت دارد. نقصان تلومر مرتبط با پروتئین Taz1 در ساکارومایسس پومبه[11] به کشیدگی زیاد تلومر و نقایص همانندسازی تلومر و نیز درهم تنیدگی های مکرر تلومر منجر میشود (Cooper و همکاران، 1997؛ Miller و Cooper، 2003). جالب اینجاست که فقدان taz1 بعلت مشکلات همانندسازی نیمه محافظه کارانه ی DNAی تلومریک به فقدان سریع تلومرها منجر می شود که با کشیدگی تلومری توسط تلومراز قابل جبران است (Miller و همکاران، 2006).
4- حذف سریع تلومر در سلولهای پستانداران
فرایندی مشابه با TRDی مخمر ابتدا در سلول های پستانداران با بیان یک آلل از پروتئین اتصالی به پروتئین TRF2 فاقد دُمین بازی ان- ترمینال: TRF2□Basic گزارش شد (Wang و همکاران، 2004). اتصال TRF2□B به تلومر ها حذف بخش های بزرگی از تکرارهای تلومریک را تحریک کرد که به کوتاه شدگی سریع تلومر می انجامد.
تکرارهای تلومریک بریده شده که دوایر تلومریک خارج کروموزومی بودند (دوایر تی[12]) مطرح میکنند که از بریدگی حلقه ی تی حاصل شدند. این حذف سریع تکرارهای تلومریک به لحاظ ژنتیکی به پروتئین های نوترکیبی همولوگ XRCC3 و NBS1 بستگی دارد. XRCC3 یک رزولواز[13] است که در جانکشن های هالیدی[14] عمل می کند (Liu و همکاران، 2004) درحالیکه NBS1 جزئی از کمپلکس MRE11-Rad50-NBS1 (MRN) است (Stracker و Petrini، 2011؛ Tauchi و همکاران، 2002). این نتایج مطرح کردند که همانند آنچه در مخمر مشاهده شد، سلولهای پستانداران قادر به بازنشانی طول تلومر ازطریق رویدادهای حذف سریع هستند.
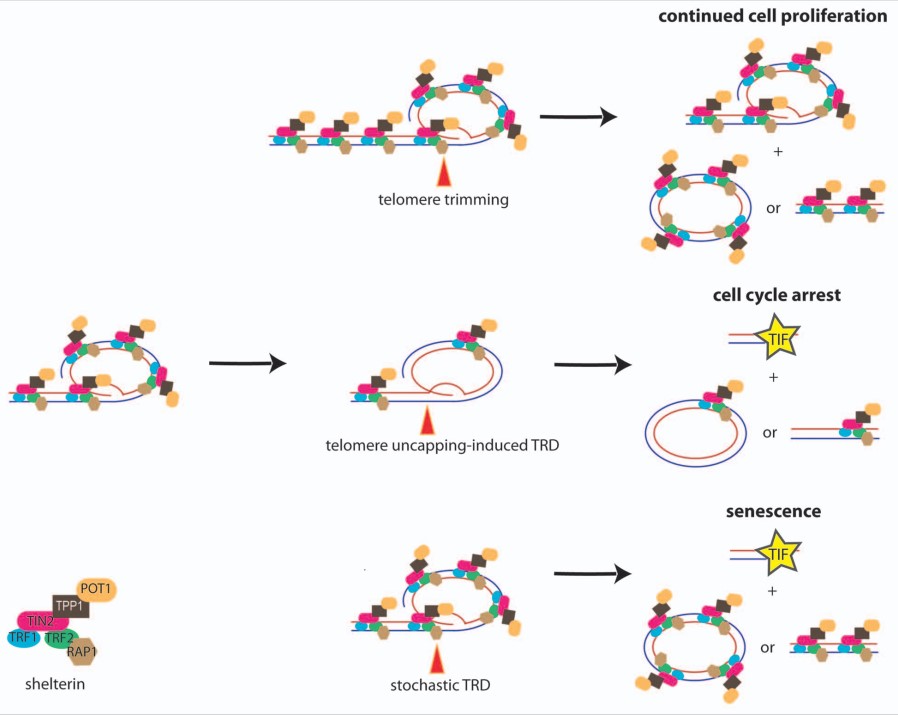
سلول های پستانداران قادر به بازنشانی طول تلومر
این اندیشه ازطریق انالیز سلول ها با تلومرهای فوق العاده کشیده اثبات شد. سطوح بالای فعالیت تلومرازی القاشده ازطریق بیش بیان همزمان جزء کاتالیزوری، hTERT، و جزء RNA، hTR، به کشیدگی پیشرونده ی تلومر منجرشد (Cristofari و Lingner، 2006).
این درحالی است که وقتی این سلول ها با سطوح بالای تلومراز در کشت حفظ شدند، کشیدگی تلومر سرانجام متوقف شد و سلول ها دوایر سی را انباشت کردند که نشانه ی نقصان سریع تلومر است (Pickett و همکاران، 2009). TRDها سبب سوء عملکرد تلومر نشدند که مطرح می کند یک فرایند تنظیم شده در تنظیم کشیدگی تلومر دخیل است، فرایندی که پیرایش تلومر نامیده شد.
فرایند مشابهی اخیرا در سلولهای بنیادی جنینی انسان (hESC) و نیز سلولهای بنیادی پرتوان القاشونده (iPCS) با سطوح تنظیم بالادستی فعالیت تلومراز تشریح شد (Rivera و همکاران، 2017). تلومرهای ابرکشیده در سلول های سرطانی انسان و نیز سلول های بنیادی پرتوان ، طول تلومر را از طریق پیرایش تلومر بازنشانی میکنند.
پیرایش تلومر در سلول های زایشی و سلول های T
پیرایش تلومر در سلول های زایشی و سلول های T برانگیخته نقش فیزیولوژیکی ایفا میکند، دو وضعیتی که فعالیت تلومراز بصورت بالادستی تنظیم میشود (Pickett و همکاران، 2011). تلومراز در سلول های زایشی در سطوح بالا قبل از لقاح برای کشیدگی تلومرها و تضمین طول مناسب تلومر در فرزند برانگیخته می شود.
القای تلومرازی در سلول های تی فعال شده برای پایایی تکثیر سلولی عظیم درطی پاسخ ایمنی لازم است. دوایر سی در هر دو وضعیت آشکار شده اند، که مطرح میکند پیرایش تلومر، مکانیسم توازن را برای جلوگیری از کشیدگی بیش ازحد تلومر تضمین می کند. دراینجا تشکیل دایره ی تی تابع XRCC3 است.
5- تفاوت بین تلومر های کوتاه و بلند چیست؟
این مشاهده که تلومرهای ابرکشیده میتوانند رویداد(های) حذف سریع را براه اندازند مطرح می کند تفاوت های بین تلومرهای کوتاه و بلند، کران بالایی طول تلومر را بازنشانی (وضع) می کند (شکل دو). یک مکانیسم بالقوه ازطریق رقیق سازی فاکتورهای ویژه ی تلومر بمجرد کشیدگی تلومر غیرطبیعی ارائه شده است.
فراوانی کمپلکس شلترین در سلول های پستانداران با تغییر طول تلومر تغییر نمی کند. درنتیجه، تراکم شلترین در تلومر ها با طول تلومر ها نسبت عکس دارد و تراکم بالاتر شلترین در تلومر های کوتاه دیده می شود (Takai و همکاران، 2010). موش با تلومر های ابرکشیده، سطوح مشابه پروتئین های شلترین در هر تلومر دارد (Varela و همکاران، 2016).
تفاوت دیگر بین تلومر های کوتاه و بلند با تغییرات کروماتین نشان داده می شود. کروماتین تلومریک در پستانداران معمولاً در حالت سرکوبگر خاموش است و توسط پروتئین 1 هتروکروماتین (HP1)، با سطوح بالای مارکر های هتروکروماتین H3K9me3 و H4K20me3 مهار می شود.
همانطور که در بالا بحث شد، کشیدگی توسط تلومراز تحت تاثیر تغییر مارکرهای کروماتین است (Benetti و همکاران، 2007؛ Gonzalo و همکاران، 2006). این داده ها مطرح می کنند که حالت هتروکروماتین، توانایی تلومراز عامل روی تلومرها را متاثر می کند و احتمالا تصور می رود که این تغییرات میتوانند واسطه ی توانایی سلول ها در مشارکت در رویداد های حذف سریع تلومر با هدف بازنشانی کشیدگی تلومر باشند.
تلومرهای طویل تر متحمل سیگنال توقف همانندسازی
داده های قوی در پشتیبانی از نقش تغییر (تعدیل) کروماتین در تنظیم کشیدگی تلومر از توصیف مسیر ALT نشات می گیرد. سلول هایی که تلومرها را با کمک این مسیر حفظ می کنند، سطوح پایین تر H3K9me3 و H4K20me3 و تراکم نوکلئوزوم پایین تر دارند (Episkopou و همکاران، 2014).
حد استانه ی بالای کشیدگی تلومر با توقف همانندسازی در تکرارهای TTAGGG تکرارشونده تحریک می شود. تلومرازها مرکب از تکرارهای TTAGGG قطاری هستند و تمایل ذاتی به تشکیل چهارتایی G دارند که بعنوان یک مانع ساختاری عمل میکند و درصورت حل نشدن، سبب فروریختن چنگال همانندسازی میشود.
درواقع DNAی تلومریک درمعرض نقایص همانندسازی است، اگر کمک پروتئین های متصل به تلومر نباشد (Miller و همکاران، 2006؛ Sfeir و همکاران، 2009). بنابراین احتمال می رود که تلومرهای طویل تر متحمل سیگنال توقف همانندسازی شوند که می تواند بواسطه ی مشارکت در حذف های تلومری سریع باشد.
القای تنش همانندسازی در تلومرها مرتبط با انباشت دوایر سی
این اندیشه با بی شمار بررسی ها پشتیبانی می شود که القای تنش همانندسازی در تلومرها را با انباشت دوایر سی مرتبط می دانند. برای نمونه، نقصان ASF1 چاپرون هسیتون مسئول اجتماع نوکلئوزومی مناسب درطی همانندسازی به انباشت دوایر سی منجر میشود (OSullivan و همکاران، 2014).
نقصان SMARCAL1 به القای دوایر سی در سلول های سرطانی مثبت تلومراز می انجامد (Poole و همکاران، 2015). SMARCAL1 عضوی از خانواده ی بازآرایی کروماتین مرتبط با SWI/SNF با فعالیت ATPase و هلیکازی است که امکان پیشروی مناسب چنگال همانندسازی درطی همانندسازی DNA را میسر می کند (Flaus و همکاران، 2006). این داده ها مطرح می کنند که توقف چنگال همانندسازی بعنوان محرک فعالیت پیرایش تلومر عمل می کند.
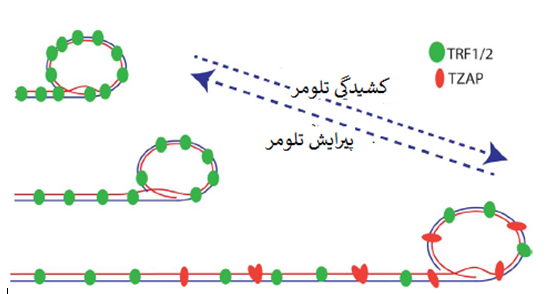
6- TZAP: پروتئین اتصالی به تلومراز در پیرایش تلومر نقش دارد
اخیرا ما و سایرین پروتئین انگشتی روی[15] ZBTB48 را بعنوان یک پروتئین اتصالی به تلومر جدید و اختصاصی شناسایی و توصیف کرده ایم. بخاطر پروتئین انگشتی روی متصل به تلومریک (TZAP) و مشخص کردن اختصاصیت این فاکتور آن را TZAP نامیدیم (Jahn و همکاران، 2017؛ Li و همکاران، 2017؛ Zhao و همکاران، 2018). TZAP بصورت مستقل از کمپلکس شلترین مستقیماً به تکرارهای TTAGGG متصل می شود.
همانطور که در سلول و آزمایشگاه نشان داده شد، اتصال TZAP به دو انتهای کروموزومی ازطریق برهم کنش مستقیم بین دُمین های انگشتی روی 3 ترمینال TZAP و تکرارهای تلومریک دورشته ای است. اتصال TZAP به تلومرها می تواند جایگزین بیش بیان TRF1 و TRF2 شود که مطرح می کند TZAP و کمپلکس شلترین برای اتصال به تلومرها رقابت دارند.
TZAP درنتیجه ی این رقابت ترجیحاً به تلومرهای طویل وصل می شود که تراکم کمپلکس شلترین درآنها کم است (Takai و همکاران، 2010) (شکل دو). استقرار TZAP در تلومرها به القای فقدان سریع توالی های تلومریک و انباشت همایند (همراه) دایره های سی منجر شد که حاکی از نقش TZAP در پیرایش تلومر است. نقصان TZAP به سطوح کاهیده ی دوایر سی و افزایش طول تلومری در ESCهای موش می انجامد (Li و همکاران، 2017).
چگونه استقرار TZAP پیرایش را افزایش می دهد؟
TZAP هیچ دمین آنزیمی ندارد، بنابراین دو سناریوی محتمل متصور است:
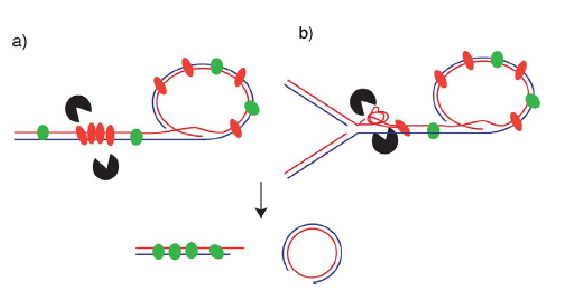
TZAP به صورت فیزیکی رزولوازها را می گیرد و با بازکردن لوپ تی سبب شکاف حلقه ی تی و پیرایش تلومر می شود (شکل سه).
کاندیداهای بالقوه عبارتند از رزولوازهای SLX4-SLX1-Mus81 یا دیسلوازهای Bloom-TopoIIIa-Rmi1. احتمال دارد که TZAP ساختارهای DNA را القا یا پایدار کند که بعنوان سوبستراهای عوامل پایین دستی عمل می کنند. TZAP در این سناریوها عمل دُمین بازی TRF2 را بی اثر می کند و در پایه ی حلقه ی تی برای جلوگیری از شکافت لوپ تی توسط رزولوازهای HJ به جانکشن های سه راهی وصل می شود (Schmutz و همکاران، 2017؛ Wang و همکاران، 2004). استانه ی بالایی طول تلومر از توازن بین اتصال TRF2 و TZAP تعیین خواهد شد.
آزمایش های بیشتر برای ارزیابی مکانیسم عمل TZAP و توانایی بالقوه اش برای اتصال و پایداری ساختارهای DNAی تلومرها لازم است.
7- پیامدهای عدم تنظیم طول تلومر
کشیدگی ناقص تلومر در همه ی سیستم های مدل تست شده نهایتاً سبب نقایص شدید رشدی/ تکثیری می شود. نقص در کشیدگی تلومر وابسته به تلومراز در انسان ها با دیسکراتوزیس مادرزادی[16] (DC) مرتبط است، اختلالی که میتواند بافت های تکثیری مختلف همچون اپیدرم و سیستم هماتوپوئتیک را درگیر کند. افراد با DC درمعرض اختلالاتی هستند که عملکرد مغز استخوان را مختل می کنند (Armanios و همکاران، 2007؛ Kirwan و Dokal، 2009؛ Shay و Wright، 1999؛ Tsakiri و همکاران، 2007).
تلومرهای بشدت کوتاه جفت شده با پاسخ نامناسب آسیب DNA علاوه بر ناهنجاری های رشدی، به تناوب چرخه های پل اتصال شکاف کمک می کنند که تومورزایی و ناپایداری ژنومی را تحریک می کنند. در واقع تلومرهای کوتاه با افزایش ریسک رشد تومورهای بافت های بسیار تکثیرشونده در مجرای روده ای، سر و گردن مرتبط هستند (Zhu و همکاران، 2016).
فنوتیپ های مشابه در موش اصلاح ژنتیکی شده ی تلومرازی گزارش شده اند که نشان دهنده ی نقص باززایی بافت و نیز آسیب پذیری افزایش یافته با گسترش سرطان است (Blasco و همکاران، 1997؛ Rudolph و همکاران، 1999). در مقابل، مفید یا مضربودن پیامدهای فیزیولوژیکی تلومرهای بسیار کشیده هنوز محل بحث است.
تلومرهای طویل و افزایش ریسک کلی سرطان
براساس این حقیقت که طول تلومر مانع تکثیر بی انتها می شود، انتظار می رود که احتمال رشد تومور با کشیدگی بیش از حد تلومر زیاد شود، بالاخص در ارگانیسم هایی که بیان تلومراز بشدت کنترل میشوند همچون پریمات ها (Gomes و همکاران، 2011).
شواهد در پشتیبانی از این اندیشه از ارتباط بین عوامل تعیین کننده ی ژنتیکی تلومرهای طویل و افزایش ریسک کلی سرطان نشات می گیرند (Rode و همکاران، 2016). تفسیر این داده ها با این واقعیت درهم تنیده است که تمایل ژنتیکی به افزایش حفظ تلومر ممکن است بجای افزایش پتانسیل تکثیر سلول های پیش از سرطانی منحصرا بعنوان مزیت بقا در سلول های سرطانی عمل کند. بررسی های انجام شده روی موش اصلاح ژنتیکی شده نشان داده اند که افزایش فعالیت تلومراز به سبب سلول های اپیتلیالی میتواند ازطریق کند کردن فرایند پیری و افزایش طول عمر مفید باشد (Tomas-Loba و همکاران، 2008).
سطوح کاهیده ی آسیب DNA و کاهش علائم پیری در موش ساخته شده از ES با تلومرهای ابرکشیده مشاهده شد (Varela و همکاران، 2016). این درحالی است که موش ها سیستم ایده آل ارزیابی تاثیر تلومرهای ابرکشیده روی رشد تومور نیستند با این فرض که کشیدگی تلومر در موش ها برای رشد تومور ضروری است (Blasco و همکاران، 1997).
[1] pluripotent
[2] telomere trimming
[3] Shelterin
[4] “t-loop
[5] ribonucleoprotein Dyskerin
[6] turnover: ترن اور
[7] regeneration رژنرسانس
[8] embryogenesis: امبریوژنز
[9] homeostasis
[10] S. cerevisiae
[11] S. pombe
[12] t-circles
[13] resolvase: آنزیمی که همواره با آنزیم تولید شده از ژن ترانسپوزاز در عناصر جابه جا شونده برای درج این عنصر در دیگر بخش های ژنوم فعالیت می کند
[14] Holliday junctions
[15] zinc finger protein
[16] dyskeratosis congenita
